Содержание
Проблемы Эволюции
Проблемы Эволюции
Предыдущая глава Следующая глава Оглавление
Доказательства эволюции
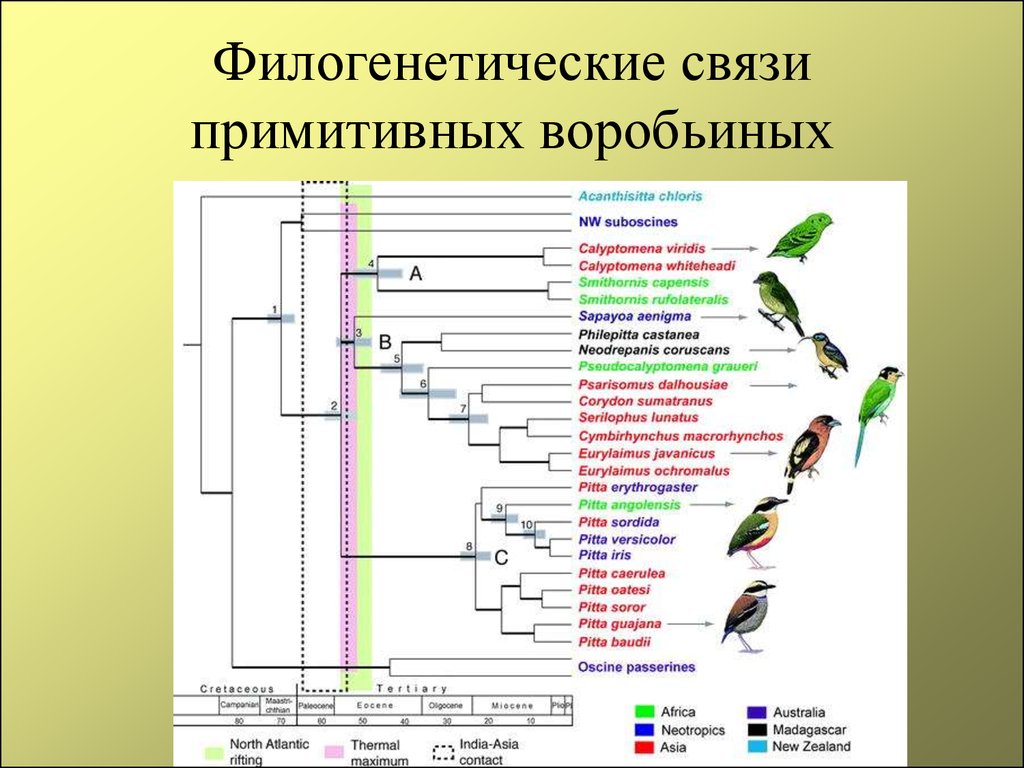
2. Эволюционное дерево
Филогенетическое дерево с указанием размера геномов.
Классификация живых организмов представляет собой многоуровневую иерархическую структуру: организмы делятся на царства, царства делятся на типы, типы — на классы, классы — на отряды, и так далее. В результате такого ветвления получается филогенетическое дерево. Наличие единственной (естественной) классификации означает, что существует объективная закономерность в основе этой классификации.Именно такой результат можно ожидать при эволюционном происхождении животных от общего предка.Ветвление филогенетического дерева соответствует делению популяций в процессе видообразования.
Несмотря на многочисленные разногласия между биологами по поводу отнесения тех или иных видов к конкретным группам (таксонам), эти противоречия имеют частный характер. Практика показывает, что биологические классификации, построенные на основе разных признаков (морфологических, эмбриологических, биохимических или генетических) в тенденции стремятся к одной и той же древовидной иерархической схеме — естественной классификации, отражающей последовательность расхождения эволюционных линий. Чем больше признаков учитывается в ходе классификации, тем выше сходство получаемых деревьев. Наличие естественной классификации было очевидно биологам еще в додарвиновские времена, и это изначально трактовалось как свидетельство иерархической организации замысла Творца. Однако в разнообразии других природных объектов, которые, в отличие от живых организмов, не происходят от общего предка, отсутствует единая древовидная иерархическая структура. Классификация таких объектов либо получается принципиально различной при использовании разных наборов признаков (например, минералы), либо имеет принципиально не «древесный» вид (например, химические элементы, звезды). Невозможно объективно построить иерархию элементарных частиц, химических элементов, планет Солнечной системы. Также не существует объективной иерархии таких сознательно созданных объектов, как книги в библиотеке, дома, мебель, машины и т. д.. Можно при желании объединять эти объекты в произвольные иерархии, но нет единственной объективной иерархии, принципиально лучшей, чем все остальные.
Невозможно объективно построить иерархию элементарных частиц, химических элементов, планет Солнечной системы. Также не существует объективной иерархии таких сознательно созданных объектов, как книги в библиотеке, дома, мебель, машины и т. д.. Можно при желании объединять эти объекты в произвольные иерархии, но нет единственной объективной иерархии, принципиально лучшей, чем все остальные.
Если бы была верна концепция «бараминов», естественная классификация и филогенетические деревья, получаемые на основе различных признаков и статистических методов, должны были бы иметь весьма характерный вид: изолированные пучки коротких ветвей (барамины) должны были бы отделяться друг от друга очень длинными (точнее, имеющими абсолютно произвольную длину) ветвями. Этого не наблюдается. Длины ветвей филогенетического дерева (отражающие, в первом приближении, степень различий по используемым признакам) в среднем примерно одинаковы между видами одного рода, родами одного семейства, семействами одного отряда и т. д. Это обстоятельство является дополнительным свидетельством в пользу того, что между процессами появления новых видов, родов, семейств, отрядов, классов, типов и т.д. не существует принципиальной разницы. Каждый новый род изначально появляется «всего лишь» как новый вид, и лишь позже, в ретроспективе, может «заслужить» в глазах биологов статус рода.
д. Это обстоятельство является дополнительным свидетельством в пользу того, что между процессами появления новых видов, родов, семейств, отрядов, классов, типов и т.д. не существует принципиальной разницы. Каждый новый род изначально появляется «всего лишь» как новый вид, и лишь позже, в ретроспективе, может «заслужить» в глазах биологов статус рода.
Кроме того, если бы в идее «бараминов» было хоть какое-то рациональное зерно, этого не могли бы не заметить специалисты по систематике животных и растений еще задолго до Дарвина. Они бы обязательно увидели, что один из надвидовых таксономических рангов (род, отряд или семейство — антиэволюционисты обычно утверждают, что их «барамины» соответствуют каким-то из этих рангов) радикально отличается от всех остальных. Например, они бы увидели, что животный мир четко делится на семейства, которые поэтому являются особым, «естественным» уровнем классификации (и соответствуют «богосотворенным родам»), и гораздо менее четко делится на роды, подсемейства, надсемейства, отряды, подклассы и т. д., которые никаким «богосотворенным родам» не соответствуют. В действительности все ранги биологической классификации имеют примерно одинаковую степень как «четкости», так и «расплывчатости». По-настоящему «естественной» единицей классификации безоговорочно признается лишь вид (по критерию полного или почти полного отсутствия скрещиваний с другими видами в природе), но барамин по определению — это не вид, а нечто большее. Однако никакого надвидового ранга, более «естественного», чем остальные, в природе не существует.
д., которые никаким «богосотворенным родам» не соответствуют. В действительности все ранги биологической классификации имеют примерно одинаковую степень как «четкости», так и «расплывчатости». По-настоящему «естественной» единицей классификации безоговорочно признается лишь вид (по критерию полного или почти полного отсутствия скрещиваний с другими видами в природе), но барамин по определению — это не вид, а нечто большее. Однако никакого надвидового ранга, более «естественного», чем остальные, в природе не существует.
Существуют различные статистические методы для точной оценки того, насколько объекты с разными свойствами укладываются в ту или иную иерархию. Эти методы измеряют так называемый «филогенетический сигнал», позволяя отличить мнимые закономерности от объективных. Например, у любого «генеалогического древа» автомобилей будет низкий уровень филогенетического сигнала. У эволюционного дерева и у различных его частей, напротив, стабильно четкий филогенетический сигнал.
Есть несколько источников данных, на основе которых можно делать выводы о степени родства между видами. Если существует единое эволюционное дерево, объединяющее все виды в объективную генеалогию, то все данные должны подтверждать эту генеалогию. Филогенетические деревья, построенные независимо, должны соответствовать друг другу. Наиболее простой, хотя и не очень точный способ построить такое дерево — сравнение строения организмов животных: чем меньше различий между видами, тем меньше поколений отделяют их от общего предка. Палеонтологическая летопись, наряду с другими подходами, позволяет установить родство между классами животных. Например, найденные останки пернатых динозавров свидетельствуют о родстве между рептилиями и птицами. Биогеография и эмбриология также дают информацию об эволюционной близости видов. Наиболее точный источник данных, недоступный во времена Дарвина — сравнительный анализ геномов различных организмов. Эволюционное дерево можно построить по каждому отдельно взятому гену, также исследователи рассматривают всевозможные некодирующие последовательности.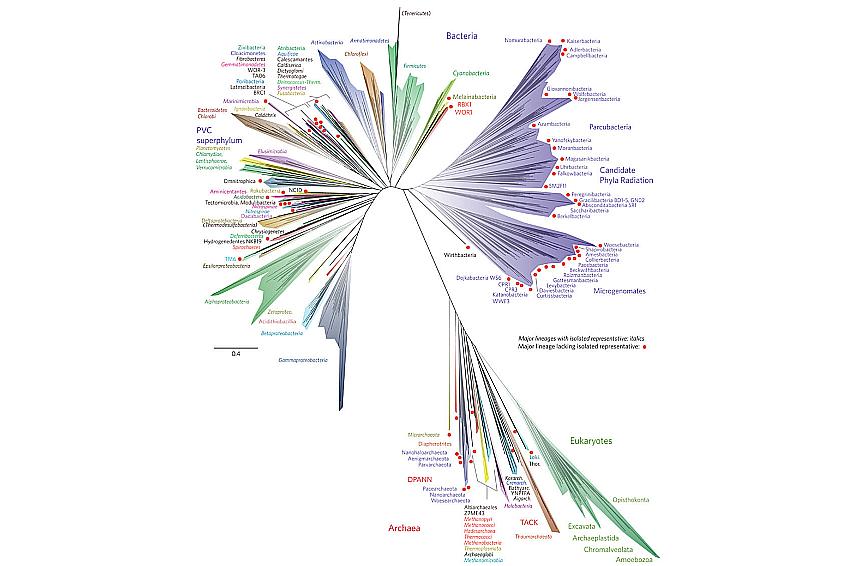 Практика показывает, что чем больше генов включается в анализ, тем меньше остается в дереве статистически слабо обоснованных участков, и тем меньше различия между деревьями, построенными по разным наборам генов.
Практика показывает, что чем больше генов включается в анализ, тем меньше остается в дереве статистически слабо обоснованных участков, и тем меньше различия между деревьями, построенными по разным наборам генов.
Все эти источники информации дают одинаковую картину с точностью до погрешности используемых методов. Тот факт, что эволюционные деревья, построенные по разным данным, соответствуют друг другу, элементарно объясняется эволюционной теорией. Другие объяснения отсутствуют: например, нет никаких причин, почему организмы, сходные по строению, должны иметь сходные некодирующие последовательности в геноме (интроны в одних и тех же местах генов, остатки встроившихся ретровирусов в одних и тех же местах генома и т.д.), если они не произошли от одного предка.
Примеры биологических исследований, связанных с построением эволюционных деревьев:
1) Новые данные позволили уточнить родословную животного царства
2) Невероятное разнообразие жуков получило эволюционное объяснение
3) Геном ланцетника помог раскрыть секрет эволюционного успеха позвоночных
4) Найдено «недостающее звено» между членистоногими и загадочными кембрийскими чудовищами аномалокарисами
5) Доказан скачкообразный характер эволюционного процесса
Горизонтальный перенос генов, межвидовая гибридизация и симбиогенез порождают горизонтальные перемычки между некоторыми ветвями эволюционного дерева, особенно в его «нижней» части — у прокариот. Это, однако, не разрушает его общую древовидную (иерархическую) структуру, потому что вертикальная передача генов (от родителей к потомкам) у всех живых организмов резко преобладает над горизонтальной.
Это, однако, не разрушает его общую древовидную (иерархическую) структуру, потому что вертикальная передача генов (от родителей к потомкам) у всех живых организмов резко преобладает над горизонтальной.
Лингвистическая эволюция как аналог биологической
Эволюция языков отчасти похожа на эволюцию биологических видов, хотя это сходство не следует преувеличивать. Лингвистическая эволюция в основном нейтральна, т.е. не имеет приспособительного характера (нельзя утверждать, например, что грамматика чукотского языка более приспособлена к холодному климату, чем грамматика африканских языков). В биологической эволюции, впрочем, тоже велик элемент нейтральности. Построение эволюционных деревьев в биологии осуществляется чаще всего на основе именно нейтральных признаков (потому что приспособительные признаки — менее надежное свидетельство родства; они часто могут возникать у неродственных форм в сходных условиях в результате одинаковой направленности отбора). Специалисты по исторической лингвистике активно и весьма успешно используют математические методы построения эволюционных деревьев, разработанные биологами (подробнее см. в заметке Лингвистическая эволюция сходна с биологической).
Специалисты по исторической лингвистике активно и весьма успешно используют математические методы построения эволюционных деревьев, разработанные биологами (подробнее см. в заметке Лингвистическая эволюция сходна с биологической).
Аналогия с эволюцией языков полезна нам прежде всего потому, что она помогает лучше понять два ключевых свойства биологической эволюции: ее постепенность и относительную дискретность видов.
Постепенность. Языки, как и биологические виды, эволюционируют путем накопления мелких изменений. Один язык (например, латынь) превращается в другой (например, итальянский) не сразу, а постепенно. Каждое следующее поколение говорит лишь немного иначе, чем предыдущее. Не бывает так, чтобы родители, говорящие, к примеру, на латыни, родили детей, которые, научившись говорить, вдруг заговорили по-итальянски. Так же и в эволюции видов: родители одного вида не могут родить детенышей, относящихся уже к другому виду. Переход между видами во времени происходит плавно и незаметно; резкие различия проявляются, только если сравнивать между собой конечные звенья длинной цепи постепенных изменений.
Переход между видами во времени происходит плавно и незаметно; резкие различия проявляются, только если сравнивать между собой конечные звенья длинной цепи постепенных изменений.
Дискретность видов. Несмотря на множество наблюдаемых в природе случаев плавных переходов между видами, разнообразных гибридных зон, кольцевых видов и т.п. (см. в разделе «Наблюдаемая эволюция»), большинство существующих видов все-таки достаточно дискретны, т.е. имеют довольно четкие границы. Обычно мы можем однозначно определить видовую принадлежность животного или растения. Между большинством видов есть заметные «разрывы» (хиатусы) в пространстве признаков. Антиэволюционисты иногда ставят это в упрек эволюционному учению, утверждая, что если бы эволюция действительно происходила, мы не должны были бы наблюдать никакой дискретности в видовом разнообразии, а только сплошные плавные переходы.
Наблюдаемая дискретность видов во многом определяется дискретностью экологических условий и пониженной приспособленностью промежуточных форм. Например, в тайге условия одни, среди арктических льдов — другие, к первым условиям лучше приспособлен бурый медведь, ко вторым — белый. Промежуточные формы в обоих биотопах будут проигрывать в конкурентной борьбе живущим там специалистам. О таком механизме формирования дискретности, основанном на пониженной конкурентоспособности промежуточных форм, писал еще Дарвин. Дополнительное объяснение дискретности можно получить из аналогии с языками.
Например, в тайге условия одни, среди арктических льдов — другие, к первым условиям лучше приспособлен бурый медведь, ко вторым — белый. Промежуточные формы в обоих биотопах будут проигрывать в конкурентной борьбе живущим там специалистам. О таком механизме формирования дискретности, основанном на пониженной конкурентоспособности промежуточных форм, писал еще Дарвин. Дополнительное объяснение дискретности можно получить из аналогии с языками.
Языки тоже в основном дискретны. Конечно, в зонах смешения разноязычных народов иногда формируются гибридные диалекты (аналог гибридных зон в биологии), но все-таки это не типично. Большинство людей говорит на вполне определенном языке, а не на каких-то смешанных диалектах. Главная причина дискретности в биологии и лингвистике, по-видимому, одна и та же. Человеку выгодно уметь свободно и полноценно обмениваться информацией с достаточно большой популяцией себе подобных. Говорить на смеси русского и немецкого очень неудобно: плохо будут понимать и русские, и немцы. Живым организмам точно так же выгодно уметь свободно и полноценно обмениваться генами (смешивая их в потомстве) с достаточно многочисленной группой особей. В пределах каждого генофонда гены под действием отбора становятся «притертыми», приспособленными друг к другу. Иметь в своем геноме смесь генов, приспособленных к разным генофондам, в общем случае невыгодно, потому что потомство такого организма от скрещивания с любой из «чистых» форм, скорее всего, будет иметь пониженную жизнеспособность. Необходимость (выгодность) свободного обмена информацией (словесной или генетической) в пределах достаточно больших популяций в тенденции приводит к формрованию и поддержанию наблюдаемой дискретности. В природных условиях довольно часто возникают межвидовые гибриды, но число их, как правило, остается небольшим, а их потомство в будущем может постепенно распасться на практически «чистых» представителей двух исходных видов. Попадая в генофонд вида А, смешанный набор генов гибридной особи будет под действием отбора постепенно очищен от «посторонних примесей», так что в итоге в генофонде останутся только гены, хорошо «приспособленные» именно к этому генофонду.
Живым организмам точно так же выгодно уметь свободно и полноценно обмениваться генами (смешивая их в потомстве) с достаточно многочисленной группой особей. В пределах каждого генофонда гены под действием отбора становятся «притертыми», приспособленными друг к другу. Иметь в своем геноме смесь генов, приспособленных к разным генофондам, в общем случае невыгодно, потому что потомство такого организма от скрещивания с любой из «чистых» форм, скорее всего, будет иметь пониженную жизнеспособность. Необходимость (выгодность) свободного обмена информацией (словесной или генетической) в пределах достаточно больших популяций в тенденции приводит к формрованию и поддержанию наблюдаемой дискретности. В природных условиях довольно часто возникают межвидовые гибриды, но число их, как правило, остается небольшим, а их потомство в будущем может постепенно распасться на практически «чистых» представителей двух исходных видов. Попадая в генофонд вида А, смешанный набор генов гибридной особи будет под действием отбора постепенно очищен от «посторонних примесей», так что в итоге в генофонде останутся только гены, хорошо «приспособленные» именно к этому генофонду. То же самое произойдет и с теми генами гибрида, которые будут подвергаться отбору в пределах генофонда Б. Именно поэтому межвидовая гибридизация, как правило, не приводит к слиянию двух разошедшихся видов в один.
То же самое произойдет и с теми генами гибрида, которые будут подвергаться отбору в пределах генофонда Б. Именно поэтому межвидовая гибридизация, как правило, не приводит к слиянию двух разошедшихся видов в один.
Предыдущая глава Следующая глава Оглавление
Филогенетическое дерево — Phylogenetic tree
Не путать с Филогинией .
Филогенетическое дерево на основе генов рРНК, показывающее три жизненных домена : бактерии, археи и эукариоты . Черная ветвь внизу филогенетического древа соединяет три ветви живых организмов с последним универсальным общим предком . В отсутствие внешней группы корень является спекулятивным.
Автоматически созданное древо жизни с высоким разрешением, основанное на полностью секвенированных геномах.
Филогенетическое дерево (также Филогения или эволюционное дерево ) представляет собой разветвленность диаграммы, или дерево, показывающее эволюционные взаимосвязи между различными биологическими видами или других объектами на основе сходства и различий в их физических или генетических характеристиках. Вся жизнь на Земле является частью единого филогенетического древа, что указывает на общее происхождение .
Вся жизнь на Земле является частью единого филогенетического древа, что указывает на общее происхождение .
В корневом филогенетическом дереве каждый узел с потомками представляет предполагаемого последнего общего предка этих потомков, а длины ребер в некоторых деревьях могут интерпретироваться как оценки времени. Каждый узел называется таксономической единицей. Внутренние узлы обычно называют гипотетическими таксономическими единицами, поскольку их нельзя наблюдать напрямую. Деревья полезны в таких областях биологии, как биоинформатика, систематика и филогенетика . Некорневые деревья иллюстрируют только родство листовых узлов и не требуют, чтобы предковый корень был известен или предполагался.
СОДЕРЖАНИЕ
- 1 История
- 2 свойства
- 2.1 Укоренившееся дерево
- 2.2 Неукорененное дерево
- 2.3 Раздвоение по сравнению с множественным
- 2.4 Маркированные в сравнении с немаркированными
- 2.5 Перечисление деревьев
- 3 особых вида деревьев
- 3.
 1 Дендрограмма
1 Дендрограмма - 3.2 Кладограмма
- 3.3 Филограмма
- 3.4 Дальгренограмма
- 3.5 Филогенетическая сеть
- 3.6 Схема шпинделя
- 3.7 Коралл жизни
- 3.
- 4 Строительство
- 4.1 Форматы файлов
- 5 Ограничения филогенетического анализа
- 6 См. Также
- 7 ссылки
- 8 Дальнейшее чтение
- 9 Внешние ссылки
- 9.1 Изображения
- 9.2 Общие
 1 Дендрограмма
1 ДендрограммаИстория
Идея « древа жизни » возникла из древних представлений о лестничном переходе от низших форм жизни к высшим (например, в Великой Цепи Бытия ). Ранние изображения «ветвящихся» филогенетических деревьев включают «палеонтологическую карту», показывающую геологические взаимоотношения между растениями и животными в книге Эдварда Хичкока « Элементарная геология » (первое издание: 1840 г.).
Чарльз Дарвин (1859) также создал одну из первых иллюстраций и чрезвычайно популяризировал понятие эволюционного «дерева» в своей основополагающей книге «Происхождение видов» . Более века спустя, эволюционные биологи до сих пор используют диаграммы дерева для описания эволюции, поскольку такие схемы эффективно передать концепцию, что видообразование происходит через адаптивный и полу случайного расщепления линий. Со временем классификация видов стала менее статичной и более динамичной.
Более века спустя, эволюционные биологи до сих пор используют диаграммы дерева для описания эволюции, поскольку такие схемы эффективно передать концепцию, что видообразование происходит через адаптивный и полу случайного расщепления линий. Со временем классификация видов стала менее статичной и более динамичной.
Термин филогенетический, или филогения, происходит от двух древнегреческих слов φῦλον ( phûlon ), означающих «раса, происхождение», и γένεσις ( génesis ), что означает «происхождение, источник».
Характеристики
Укоренившееся дерево
Укоренившееся филогенетическое дерево, оптимизированное для слепых. Самая низкая точка дерева — корень, который символизирует универсального общего предка всех живых существ. Дерево разветвляется на три основные группы: бактерии (левая ветвь, буквы от a до i), археи (средняя ветвь, буквы от j до p) и эукариоты (правая ветвь, буквы от q до z). Каждая буква соответствует группе организмов, перечисленных под этим описанием. Эти буквы и описание следует преобразовать в шрифт Брайля и распечатать на принтере Брайля. Фигуру можно напечатать на 3D-принтере, скопировав файл png и используя Cura или другое программное обеспечение для генерации G-кода для 3D-печати.
Эти буквы и описание следует преобразовать в шрифт Брайля и распечатать на принтере Брайля. Фигуру можно напечатать на 3D-принтере, скопировав файл png и используя Cura или другое программное обеспечение для генерации G-кода для 3D-печати.
Филогенетическое дерево с корнем (см. Два рисунка вверху) — это ориентированное дерево с уникальным узлом — корнем — соответствующим (обычно условно исчисляемому ) самому последнему общему предку всех сущностей на листьях дерева. Корневой узел не имеет родительского узла, но служит родительским для всех остальных узлов в дереве. Таким образом, корень является узлом степени 2, в то время как другие внутренние узлы имеют минимальную степень 3 (где «степень» здесь относится к общему количеству входящих и исходящих ребер).
Наиболее распространенный метод укоренения деревьев — использование непротиворечивой внешней группы — достаточно тесной, чтобы можно было сделать вывод на основании данных о признаках или молекулярного секвенирования, но достаточно далеко, чтобы быть чистой внешней группой.
Неукорененное дерево
Некорневое филогенетическое дерево для миозина, A надсемейство из белков .
Некорневые деревья иллюстрируют родство листовых узлов без каких-либо предположений о происхождении. Они не требуют, чтобы предковый корень был известен или предполагался. Некорневые деревья всегда можно сгенерировать из корневых, просто опуская корень. Напротив, определение корня дерева без корней требует некоторых средств определения происхождения. Обычно это делается путем включения внешней группы во входные данные, чтобы корень обязательно находился между внешней группой и остальными таксонами в дереве, или путем введения дополнительных предположений об относительной скорости эволюции в каждой ветви, например, приложения от молекулярных часов гипотезы .
Раздвоение против множественного
И корневые, и неукорененные деревья могут быть как разветвляющимися, так и множественными. У корневого бифуркационного дерева есть ровно два потомка, возникающих из каждого внутреннего узла (то есть оно образует бинарное дерево ), а бифуркационное дерево без корня принимает форму бифуркационного дерева без корня, свободного дерева с ровно тремя соседями в каждом внутреннем узле. Напротив, у корневого мультифуркационного дерева может быть более двух дочерних узлов на некоторых узлах, а у некорневого мультифуркационного дерева может быть более трех соседей на некоторых узлах.
Напротив, у корневого мультифуркационного дерева может быть более двух дочерних узлов на некоторых узлах, а у некорневого мультифуркационного дерева может быть более трех соседей на некоторых узлах.
Маркированные или немаркированные
И деревья с корнями, и деревья без корней могут быть помечены или немаркированы. Помеченное дерево имеет определенные значения, присвоенные его листьям, в то время как немаркированное дерево, иногда называемое формой дерева, определяет только топологию. Некоторые деревья на основе последовательностей, построенные из небольшого геномного локуса, такие как Phylotree, имеют внутренние узлы, помеченные предполагаемыми предковыми гаплотипами.
Перечисление деревьев
Увеличение общего количества филогенетических деревьев в зависимости от количества помеченных листьев: бинарные деревья без корней (синие ромбы), бинарные деревья с корнями (красные кружки) и многоцветные или бинарные деревья с корнями (зеленые: треугольники). {6}}
{6}}
| Маркированные листья | Бинарные некорневые деревья | Бинарные корневые деревья | Многоплодные корневые деревья | Все возможные корневые деревья |
|---|---|---|---|---|
| 1 | 1 | 1 | 0 | 1 |
| 2 | 1 | 1 | 0 | 1 |
| 3 | 1 | 3 | 1 | 4 |
| 4 | 3 | 15 | 11 | 26 год |
| 5 | 15 | 105 | 131 | 236 |
| 6 | 105 | 945 | 1 807 | 2 752 |
| 7 | 945 | 10 395 | 28 813 | 39 208 |
| 8 | 10 395 | 135 135 | 524 897 | 660 032 |
| 9 | 135 135 | 2 027 025 | 10,791,887 | 12 818 912 |
| 10 | 2 027 025 | 34 459 425 | 247 678 399 | 282 137 824 |
Особые виды деревьев
Дендрограмма филогении некоторых пород собак
Дендрограмма
Дендрограммы это общее название для дерева, будь то филогенетического или нет, а следовательно, и на схематическом представлении филогенетического дерева.
Кладограмма
Кладограмма представляет собой лишь ветвление; т.е. длины его ветвей не представляют время или относительную величину изменения символа, а его внутренние узлы не представляют предков.
Хронограмма чешуекрылых . В этом филогенетическом типе деревьев длина ветвей пропорциональна геологическому времени.
Филограмма
Филограмма — это филогенетическое дерево, длина ветвей которого пропорциональна изменению характера.
Хронограмма — это филогенетическое дерево, которое явно представляет время по длине ветвей.
Дальгренограмма
Dahlgrenogram является схемой, представляющей поперечное сечение филогенетического дерева
Филогенетическая сеть
Филогенетическое сеть строго говоря, не дерево, а более общий график, или ориентированный ациклический граф в случае укорененных сетей. Они используются для преодоления некоторых ограничений, присущих деревьям.
Схема шпинделя
Диаграмма веретена, показывающая эволюцию позвоночных на уровне класса, ширина веретена указывает количество семейств. Диаграммы шпинделя часто используются в эволюционной систематике .
Диаграммы шпинделя часто используются в эволюционной систематике .
Диаграмму веретена или пузырьковую диаграмму часто называют ромерограммой после ее популяризации американским палеонтологом Альфредом Ромером . Он представляет таксономическое разнообразие (горизонтальная ширина) в зависимости от геологического времени (вертикальная ось), чтобы отразить изменение численности различных таксонов во времени. Однако диаграмма веретена не является эволюционным деревом: таксономические веретена скрывают фактические отношения родительского таксона с дочерним таксоном и имеют недостаток, заключающийся в вовлечении парафилии родительской группы. Этот тип диаграммы больше не используется в первоначально предложенной форме.
Коралл жизни
Коралл жизни
Дарвин также упомянул, что коралл может быть более подходящей метафорой, чем дерево . Действительно, филогенетические кораллы полезны для изображения прошлой и настоящей жизни, и у них есть некоторые преимущества перед деревьями (разрешены анастомозы и т. Д.).
Д.).
Строительство
Основная статья: Вычислительная филогенетика
Филогенетические деревья, состоящие из нетривиального числа входных последовательностей, строятся с использованием методов вычислительной филогенетики . Методы Расстояние-матрицы, такие как сосед-присоединения или UPGMA, которые вычисляют генетическое расстояние от множественного выравнивания последовательностей, наиболее просты в реализации, но не вызывают эволюционной модели. Многие методы выравнивания последовательностей, такие как ClustalW, также создают деревья с помощью более простых алгоритмов (то есть основанных на расстоянии) построения дерева. Максимальная экономия — еще один простой метод оценки филогенетических деревьев, но подразумевает неявную модель эволюции (то есть экономию). Более продвинутые методы используют критерий оптимальности по максимальной вероятности, часто в рамках байесовского и применить явную модель эволюции оценки филогенетического дерева. Идентификация оптимального дерева с использованием многих из этих методов является NP-сложной задачей, поэтому методы эвристического поиска и оптимизации используются в сочетании с функциями оценки дерева для определения достаточно хорошего дерева, которое соответствует данным.
Методы построения деревьев можно оценить по нескольким критериям:
- эффективность (сколько времени нужно, чтобы вычислить ответ, сколько памяти ему нужно?)
- мощность (хорошо ли используются данные или информация тратится зря?)
- согласованность (будет ли он постоянно сходиться к одному и тому же ответу, если каждый раз давать разные данные для одной и той же модельной задачи?)
- устойчивость (хорошо ли справляется с нарушениями предположений базовой модели?)
- фальсифицируемость (предупреждает ли она нас о том, что использование нецелесообразно, т.е. когда предположения нарушаются?)
Методы построения деревьев также привлекли внимание математиков. Деревья также могут быть построены с использованием Т-теории .
Форматы файлов
Деревья могут быть закодированы в различных форматах, каждый из которых должен представлять вложенную структуру дерева. Они могут кодировать или не кодировать длины ветвей и другие особенности. Стандартизированные форматы критически важны для распространения и совместного использования деревьев, не полагаясь на вывод графики, который трудно импортировать в существующее программное обеспечение. Обычно используемые форматы:
Стандартизированные форматы критически важны для распространения и совместного использования деревьев, не полагаясь на вывод графики, который трудно импортировать в существующее программное обеспечение. Обычно используемые форматы:
- Формат файла Nexus
- Формат Ньюика
Ограничения филогенетического анализа
Хотя филогенетические деревья, созданные на основе секвенированных генов или геномных данных у разных видов, могут дать представление об эволюции, этот анализ имеет важные ограничения. Что наиболее важно, деревья, которые они производят, не обязательно правильные — они не обязательно точно отражают эволюционную историю включенных таксонов. Как и любой научный результат, они могут быть фальсифицированы путем дальнейшего изучения (например, сбора дополнительных данных, анализа существующих данных улучшенными методами). Данные, на которых они основаны, могут быть зашумленными ; анализ может быть затруднен генетической рекомбинацией, горизонтальным переносом генов, гибридизацией между видами, которые не были ближайшими соседями на дереве до того, как произошла гибридизация, конвергентной эволюцией и консервативными последовательностями .
Кроме того, существуют проблемы с основанием анализа на одном типе признака, таком как один ген или белок, или только на морфологическом анализе, потому что такие деревья, построенные из другого несвязанного источника данных, часто отличаются от первого, и поэтому требуется большая осторожность. в выводе филогенетических отношений между видами. Это наиболее верно в отношении генетического материала, который подвержен латеральному переносу генов и рекомбинации, где разные блоки гаплотипов могут иметь разную историю. В этих типах анализа выходное дерево филогенетического анализа отдельного гена является оценкой филогении гена (т. Е. Генного дерева), а не филогении таксонов (т. Е. Дерева видов), из которых были взяты эти признаки, хотя в идеале оба должны быть очень близко. По этой причине серьезные филогенетические исследования обычно используют комбинацию генов, происходящих из разных геномных источников (например, из митохондриальных или пластидных или ядерных геномов), или генов, которые, как ожидается, будут развиваться при различных режимах отбора, так что гомоплазия (ложная гомология ) вряд ли возникнет в результате естественного отбора.
Когда вымершие виды включаются в анализ в качестве конечных узлов (а не, например, для ограничения внутренних узлов), считается, что они не представляют прямых предков каких-либо существующих видов. Вымершие виды обычно не содержат ДНК высокого качества .
Диапазон полезных материалов ДНК расширился с развитием технологий экстракции и секвенирования. Разработка технологий, способных выводить последовательности из более мелких фрагментов или из пространственных структур продуктов деградации ДНК, еще больше расширит диапазон ДНК, считающихся полезными.
Филогенетические деревья также могут быть выведены из ряда других типов данных, включая морфологию, наличие или отсутствие определенных типов генов, события вставки и удаления — и любые другие наблюдения, которые, как считается, содержат эволюционный сигнал.
Филогенетические сети используются, когда разветвляющиеся деревья не подходят из-за этих сложностей, которые предполагают более сетчатую эволюционную историю отобранных организмов.
Смотрите также
- Портал эволюционной биологии
- Clade
- Кладистика
- Вычислительная филогенетика
- Эволюционная биология
- Эволюционная таксономия
- Обобщенное выравнивание дерева
- Список программ филогенетики
- Список программ визуализации филогенетического дерева
- PANDIT, биологическая база данных, охватывающая белковые домены
- Филогенетические сравнительные методы
использованная литература
дальнейшее чтение
- Schuh, RT и AVZ Brower. 2009. Биологическая систематика: принципы и приложения (2-е изд.) ISBN 978-0-8014-4799-0
- Мануэль Лима, Книга деревьев: визуализация отраслей знания, 2014, Princeton Architectural Press, Нью-Йорк.
- MEGA, бесплатная программа для рисования филогенетических деревьев.
- Гонтье, Н. 2011. «Изображение Древа Жизни: философские и исторические корни эволюционных древовидных диаграмм». Эволюция, образование, просветительская деятельность 4: 515–538.

внешние ссылки
Изображений
- Y-хромосома человека 2002 Филогенетическое дерево
- iTOL: Интерактивное Древо Жизни
- Филогенетическое древо искусственных организмов, эволюционировавших на компьютерах
- Филограмма эвтерианских млекопитающих Миямото и Гудмана
Общий
- Обзор различных методов визуализации дерева доступен на странице Page, RDM (2011). «Пространство, время, форма: просмотр Древа Жизни». Тенденции в экологии и эволюции . 27 (2): 113–120. DOI : 10.1016 / j.tree.2011.12.002 . PMID 22209094 .
- OneZoom: Tree of Life — все живые виды в виде интуитивно понятного и масштабируемого исследователя фракталов (адаптивный дизайн)
- Откройте для себя жизнь Интерактивное дерево на основе проекта Национального научного фонда США «Сборка дерева жизни».
- Филокод
- Множественное совмещение 139 последовательностей миозина и филогенетического дерева
- Веб-проект «Древо жизни»
- Филогенетический вывод на сервере T-REX
- База данных таксономии NCBI [1]
- ETE: среда Python для исследования деревьев. Это программная библиотека для анализа, управления и визуализации филогенетических деревьев. Ref.
- Ежедневно обновляемое дерево (упорядоченной) жизни Fang, H .; Оутс, Мэн; Петика, РБ; Гринвуд, JM; Сардар, AJ; Rackham, OJL; Донохью, PCJ; Stamatakis, A .; Де Лима Мораис, DA; Гоф, Дж. (2013). «Ежедневно обновляемое дерево (секвенированной) жизни как справочник для исследования генома» . Научные отчеты . 3 : 2015. Bibcode : 2013NatSR … 3E2015F . DOI : 10.1038 / srep02015 . PMC 6504836 . PMID 23778980 .
 Это программная библиотека для анализа, управления и визуализации филогенетических деревьев. Ref.
Это программная библиотека для анализа, управления и визуализации филогенетических деревьев. Ref.<img src=»//en.wikipedia.org/wiki/Special:CentralAutoLogin/start?type=1×1″ alt=»»>
Генетики «редактируют» родословное древо птиц
Никита Зеленков
«Наука из первых рук» №1(61), 2015
С момента зарождения теории эволюции Дарвина ученые пытаются воссоздать древо жизни для всех ныне живущих на земле организмов, и именно птицы долгое время «задавали тон» в подобных исследованиях. Однако древо, отражающее представления ученых конца XIX в. о родственных отношениях птиц, мало менялось в течение последующих десятилетий, а в некоторых странах (например, в России) орнитологи предпочитают придерживаться его и поныне. Ситуация в корне переменилась в 1990 г., когда американские орнитологи Ч. Сибли и Д. Олквист построили совершенно новое филогенетическое древо птиц исключительно на основе сходства их ДНК, не оставив и следа от традиционной классификации птиц. С этого времени отмечается всевозрастающий интерес к проблеме родственных отношений между птицами. Однако до последнего времени все работы базировались на поиске сходства последовательностей нуклеотидов в молекулах ядерной ДНК. И только в 2014 г. в Science было опубликовано новое филогенетическое древо, построенное на основе исследования полного генома у представителей всех отрядов птиц. Согласно новому древу, например, ближайшими родственниками фламинго и поганок в нашей фауне могут оказаться голуби, а ближайшими родственниками дроф — кукушки. Но особенно важно то, что теперь мы знаем, что современные представления о родстве между птицами базируются на основе сходства не только между отдельными генами и их комбинациями, но и всего генома.
о родственных отношениях птиц, мало менялось в течение последующих десятилетий, а в некоторых странах (например, в России) орнитологи предпочитают придерживаться его и поныне. Ситуация в корне переменилась в 1990 г., когда американские орнитологи Ч. Сибли и Д. Олквист построили совершенно новое филогенетическое древо птиц исключительно на основе сходства их ДНК, не оставив и следа от традиционной классификации птиц. С этого времени отмечается всевозрастающий интерес к проблеме родственных отношений между птицами. Однако до последнего времени все работы базировались на поиске сходства последовательностей нуклеотидов в молекулах ядерной ДНК. И только в 2014 г. в Science было опубликовано новое филогенетическое древо, построенное на основе исследования полного генома у представителей всех отрядов птиц. Согласно новому древу, например, ближайшими родственниками фламинго и поганок в нашей фауне могут оказаться голуби, а ближайшими родственниками дроф — кукушки. Но особенно важно то, что теперь мы знаем, что современные представления о родстве между птицами базируются на основе сходства не только между отдельными генами и их комбинациями, но и всего генома.
Второе место среди десятки самых значительных научных достижений 2014 г. редакторы журнала Science отдали работам большого коллектива эволюционных биологов, которые с помощью современных молекулярно-генетических методов провели «ревизию» родословной птиц — наиболее многочисленного и широко распространенного класса современных позвоночных, — оценив темп и направление эволюционных переходов внутри этой группы на основе анализа геномов представителей разных таксонов пернатых.
Птицы на протяжении столетий привлекали особое внимание исследователей, поэтому неудивительно, что многие открытия в различных областях биологической науки были сделаны при исследовании именно этих высших позвоночных. Вспомним, что даже в формировании теории происхождения видов важнейшую роль сыграли галапагосские вьюрки, так что этих птиц теперь по праву называют «дарвиновыми». Эти небольшие певчие птицы, относящиеся к танагровым — семейству всеядных птиц Нового Света, не так давно заселили Галапагосский архипелаг, где успели «произвести» на свет более десяти видов, различающихся размером, окраской оперения и строением клюва. Во время своего знаменитого путешествия на корабле «Бигль» Чарльз Дарвин посетил Галапагосы, где и наблюдал удивительное разнообразие этих птиц, являющееся более чем наглядной и убедительной иллюстрацией их недавней эволюции (по современным оценкам, предок современных галапагосских вьюрков попал на острова не ранее 2–3 млн лет назад).
Во время своего знаменитого путешествия на корабле «Бигль» Чарльз Дарвин посетил Галапагосы, где и наблюдал удивительное разнообразие этих птиц, являющееся более чем наглядной и убедительной иллюстрацией их недавней эволюции (по современным оценкам, предок современных галапагосских вьюрков попал на острова не ранее 2–3 млн лет назад).
Теория эволюции Дарвина оказала важнейшие влияние на развитие современной науки. Одним из существенных ее следствий стало понимание, что жизнь на Земле можно представить в виде своеобразного древа — что ныне живущие виды происходят от каких-то вымерших предков, а те, в свою очередь, восходят к каким-то еще более далеким предкам. По мере удаления в прошлое «ветви», представленные современными видами, сливаются во все более толстые «сучья» и заканчиваются единым «стволом» — предполагаемым предком всего живого на Земле. Очень примечательно, что именно такое дерево жизни, которое можно уподобить генеалогическому дереву человеческого рода, было единственной иллюстрацией в первом издании знаменитой книги Ч. Дарвина «Происхождение видов…».
Дарвина «Происхождение видов…».
С момента зарождения теории эволюции Дарвина ученые пытаются воссоздать древо жизни для всех ныне живущих на земле организмов, и именно птицы долгое время, что называется, «задавали тон» в подобных исследованиях. Во второй половине XIX в. материал для реконструкции такого древа жизни (или, на современном языке, — «филогенетического древа») ученые черпали во внутреннем строении организмов. Уже Дарвину было понятно, что внешнее строение животных может меняться в зависимости от условий среды, при этом животные, приспосабливаясь к сходным условиям, могут стать удивительно похожими друг на друга. В этом смысле хорошим примером являются такие рыбы, как акулы, и касатки — водные млекопитающие.
В XIX в. считалось, что анатомия внутренних органов гораздо меньше или даже совсем не зависит от условий среды, и поэтому именно она годится для выявления родственных связей между организмами. С этой целью была исследована мускулатура, кости, кровеносная система и некоторые другие системы органов различных видов птиц. На основе сходства внутреннего строения и благодаря усилиям ряда ученых к концу этого столетия удалось построить вполне сносное филогенетическое древо птиц, которое более или менее исправно служило биологам и на протяжении большей части XX в. Самую заметную роль в создании базисных идей о взаимном родстве птиц сыграл немецкий анатом М. Фюрбрингер, издавший в 1888 г. поистине гигантский фолиант по сравнительной анатомии птиц, содержавшей свыше 1700 страниц современного формата A3 (Fürbringer, 1888).
На основе сходства внутреннего строения и благодаря усилиям ряда ученых к концу этого столетия удалось построить вполне сносное филогенетическое древо птиц, которое более или менее исправно служило биологам и на протяжении большей части XX в. Самую заметную роль в создании базисных идей о взаимном родстве птиц сыграл немецкий анатом М. Фюрбрингер, издавший в 1888 г. поистине гигантский фолиант по сравнительной анатомии птиц, содержавшей свыше 1700 страниц современного формата A3 (Fürbringer, 1888).
Нужно сказать, что древо, отражающее представления ученых конца XIX в. о родственных отношениях птиц, мало менялось в течение последующих десятилетий, а в некоторых странах (например, в России) орнитологи предпочитают придерживаться его и поныне. Откройте любой определитель птиц на русском языке, и вы убедитесь, что он начинается с поганок и гагар — птиц, хорошо приспособленных к водному образу жизни и поэтому похожих, но далеко не близкородственных. Хотя уже в 1970–1980-х гг. стало ясно, что в традиционных представлениях о родстве между птицами имеются многочисленные ошибки, что внутренняя анатомия также подвержена параллельной эволюции, поэтому сходство между птицами может не отражать их происхождение от недавнего общего предка.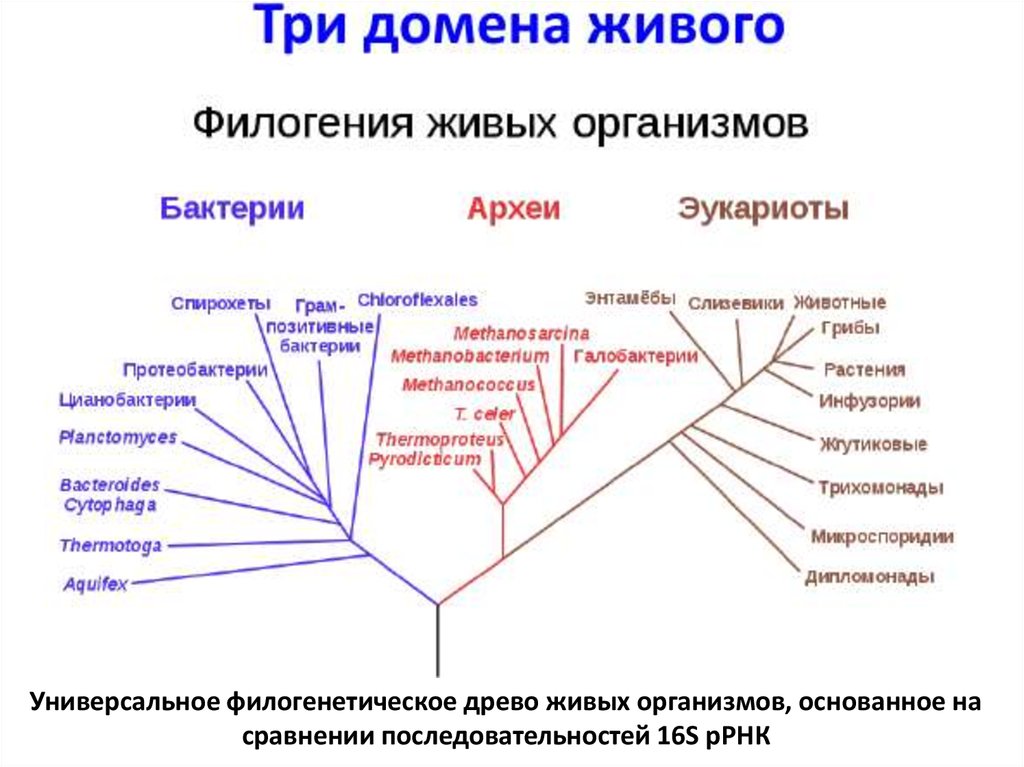
Одними из первых на недостатки традиционной классификации птиц обратили внимание, пожалуй, палеонтологи, изучавшие вымерших представителей. Анатомы, которые к этому времени начали исследовать внутреннее строение животных совершенно на другом уровне, также стали задаваться вопросами о родственных отношениях птиц между собой. Отдельные ученые предпринимали попытки улучшить птичье филогенетическое древо, однако эти усилия не имели особого успеха — во многом потому, что орнитологическое сообщество упорно не хотело пересматривать традиционные представления о родстве пернатых между собой.
Ситуация в корне переменилась в 1990 г., когда была опубликована эпохальная книга американских орнитологов Ч. Сибли и Д. Олквиста, посвященная молекулярной эволюции птиц (Sibley, Ahlquist, 1990), — первая в истории науки работа, охватившая молекулярную эволюцию крупной группы животных. Авторы книги построили совершенно новое филогенетическое древо птиц исключительно на основе сходства их ДНК, при этом от традиционной классификации птиц не осталось и следа. Например, на удивление всех орнитологов, ближайшими родичами певчих птиц выступили голуби, журавли и аисты, но вовсе не дятлы, как это считалось ранее. Зато дятлы и туканы вдруг оказались представителями одной из самых древних независимых линий, при том что они и внешне, и по внутреннему строению очень похожи на певчих птиц.
Например, на удивление всех орнитологов, ближайшими родичами певчих птиц выступили голуби, журавли и аисты, но вовсе не дятлы, как это считалось ранее. Зато дятлы и туканы вдруг оказались представителями одной из самых древних независимых линий, при том что они и внешне, и по внутреннему строению очень похожи на певчих птиц.
Новая классификация птиц Сибли и Олквиста была встречена очень резкой критикой — во многом из-за проблем с методологией. Время показало, что это филогенетическое древо действительно было во многом ошибочным, однако его значение для развития науки об эволюции птиц трудно переоценить. Именно эта работа заставила рядовых орнитологов допустить саму мысль о том, что традиционные представления о родственных отношениях между птицами могут быть в принципе неверными.
С середины 1990-х гг. отмечается всевозрастающий интерес к проблеме родственных отношений между птицами. По этой теме ежегодно публикуются десятки работ, благодаря чему у нас постепенно выстраивается все более ясная картина нового древа жизни птиц. Какие-то из традиционных воззрений подтверждаются, какие-то полностью отвергаются. При этом на первое место по значимости выходят данные анализа последовательности нуклеотидов в молекулах ядерной ДНК (не просто сходство, как у Сибли и Олквиста, а «продвинутое» сходство).
Какие-то из традиционных воззрений подтверждаются, какие-то полностью отвергаются. При этом на первое место по значимости выходят данные анализа последовательности нуклеотидов в молекулах ядерной ДНК (не просто сходство, как у Сибли и Олквиста, а «продвинутое» сходство).
Исследования отдельных генов и редких геномных изменений позволили выявить много нового и порою неожиданного. Так, в 2001 г. было обнаружено, что ближайшими родственниками поганок являются вовсе не гагары, так на них похожие, а фламинго, с которыми у поганок, казалось бы, нет ничего общего! Поначалу сближение поганок и фламинго представлялось какой-то ошибкой молекулярных биологов, однако исследования все новых и новых генов подтверждали их близкое родство. Позже выяснилось, что у поганок и фламинго на пальцах ног имеются ногти, а у всех остальных птиц — когти, что добавило уверенности в правомерности сближения этих птиц. В конце концов удалось найти и давно вымершую группу птиц — промежуточную между фламинго и поганками. Ими оказались ископаемые фламинго палелодиды, которые в отличие от современных были очень хорошо приспособлены к плаванию и, возможно, даже к нырянию (хотя современные фламинго в принципе могут плавать, однако они мало адаптированы к такому занятию).
Ими оказались ископаемые фламинго палелодиды, которые в отличие от современных были очень хорошо приспособлены к плаванию и, возможно, даже к нырянию (хотя современные фламинго в принципе могут плавать, однако они мало адаптированы к такому занятию).
В результате тщательнейших исследований, проведенных множеством специалистов за последние 15 лет, представления о древе жизни птиц были полностью переработаны. Если бы мы сегодня создавали новый определитель птиц России, то должны были бы начинать его уже не с гагар и поганок, а с гусей, уток и фазанов, которые оказались близкими родственниками и самыми примитивными из наших птиц. Поганки и фламинго, о которых шла речь выше, могли бы идти следом. Ближайшими родственникам певчих птиц могут быть либо попугаи, либо соколы. А вот ястребы, которые раньше считались родственниками соколов, теперь помещаются совсем в другую эволюционную ветвь. Уже нет сомнений, что стрижи и колибри родственны козодоям, а цапли — не аистам, а веслоногим (например, пеликанам). Что дрофы — это отдельная линия птиц, не родственная журавлеобразным, как считалось ранее. Такую новую классификацию птиц, учитывающую современные достижения молекулярной биологии и палеонтологии, автор предложил в позапрошлом году (Зеленков, 2013).
Что дрофы — это отдельная линия птиц, не родственная журавлеобразным, как считалось ранее. Такую новую классификацию птиц, учитывающую современные достижения молекулярной биологии и палеонтологии, автор предложил в позапрошлом году (Зеленков, 2013).
Хотя мы сейчас, несомненно, знаем о родстве между различными группами птиц намного больше, чем раньше, неясного осталось еше предостаточно. Например, не до конца понятно, какое положение на филогенетическом древе птиц занимают голуби, совы, дрофы и др. В настоящее время считается, что ответить на ряд вопросов поможет исследование всего генома (до сих пор исследовались только отдельные гены либо относительно небольшие «порции» генома).
В связи с этим так важны результаты геномного исследования филогенетического дерева птиц, опубликованные в одном из недавних выпусков Science (Jarvis et al., 2014). Авторы этой работы — очень большой коллектив ученых, исследовали полный геном у представителей всех отрядов птиц и на этом основании построили новое филогенетическое древо, которое можно назвать очередным существенным шагом вперед к пониманию родства между современными пернатыми. Нужно отметить, что полученное авторами филогенетическое дерево предоставило очень мало сюрпризов: большинство обнаруженных ими ветвей так или иначе уже были выявлены в последние десятилетия (например, те же поганки и фламинго). Но есть и новые интересные результаты: например, ближайшими родственниками фламинго и поганок в нашей фауне могут оказаться голуби, а ближайшими родственниками дроф — кукушки.
Нужно отметить, что полученное авторами филогенетическое дерево предоставило очень мало сюрпризов: большинство обнаруженных ими ветвей так или иначе уже были выявлены в последние десятилетия (например, те же поганки и фламинго). Но есть и новые интересные результаты: например, ближайшими родственниками фламинго и поганок в нашей фауне могут оказаться голуби, а ближайшими родственниками дроф — кукушки.
Особенно важно то, что теперь мы знаем, что современные представления о родстве между птицами базируются на основе сходства не только между отдельными генами и их комбинациями, но и всего генома. Дальнейшее развитие этого направления видится в изучении все большего числа геномов от птиц разных видов, ведь именно от этого во многом будет зависеть форма ветвления полученного филогенетического древа. Можно утверждать, что несмотря на долгий прогресс в науке об эволюции птиц, мы все еще стоим на пороге больших открытий.
Литература:
1. Зеленков Н. В. Система птиц (Aves: Neornithes) в начале XXI века // Труды Зоологического ин-та РАН. 2013. Прил. № 2. С. 174–190.
2013. Прил. № 2. С. 174–190.
2. Jarvis E. D. et al. Whole-genome analyses resolve early branches in the tree of life of modern birds // Science. 2014. V. 346. № 6215. P. 1320–1331.
3. Sibley C. G., Ahlquist J. E. Phylogeny and classification of birds: a study in molecular evolution. Yale University Press, New Heaven, London, 1990. 976 p.
филогенетических деревьев | Биологические принципы
Цели обучения
- Знать и использовать терминологию, необходимую для описания и интерпретации филогенетического дерева.
- Знать различные типы данных, включенных в филогенетические деревья, и понимать, как эти данные используются для построения филогенетических деревьев
- Интерпретация родства существующих видов на основе филогенетических деревьев
Что такое филогенетическое дерево?
Филогенетическое древо — это визуальное представление взаимоотношений между различными организмами, показывающее путь во времени эволюции от общего предка к разным потомкам. Деревья могут представлять отношения, начиная от всей истории жизни на земле и заканчивая отдельными людьми в популяции.
Деревья могут представлять отношения, начиная от всей истории жизни на земле и заканчивая отдельными людьми в популяции.
На приведенной ниже диаграмме показано дерево из 3 таксонов (отдельный таксон является таксономической единицей; может быть видом или геном).
Терминология филогенетических деревьев
Это разветвляющееся дерево. Вертикальные линии, называемые ветвями , представляют родословную , а узла находятся там, где они расходятся, представляя событие видообразования от общего предка. Ствол у основания дерева на самом деле называется корнем . Корневой узел представляет самый последний общий предок всех таксонов, представленных на дереве. Время также представлено, начиная от самого старого внизу до самого последнего вверху. Это конкретное дерево говорит нам о том, что таксон A и таксон B более тесно связаны друг с другом, чем любой таксон с таксоном C. Причина в том, что таксон A и таксон B имеют более позднего общего предка, чем таксон C. Группа таксонов, включающая общего предка и всех его потомков, называется таксонов.0023 клада . Также говорят, что клада монофилетическая . Группа, которая исключает одного или нескольких потомков , является парафилетической ; группа, исключающая общего предка r, называется полифилетической.
Группа таксонов, включающая общего предка и всех его потомков, называется таксонов.0023 клада . Также говорят, что клада монофилетическая . Группа, которая исключает одного или нескольких потомков , является парафилетической ; группа, исключающая общего предка r, называется полифилетической.
На изображении ниже показаны несколько монофилетических (верхний ряд) и полифилетических (внизу слева) или парафилетических (внизу справа) деревьев. Обратите внимание, что клады включают общего предка и всех его потомков (зеленый и синий примеры), в то время как клады, помеченные как «не клада», не включают некоторых общих предков (полифилетические, выделенные красным) или некоторых потомков (парафилетические, выделенные оранжевым).
Взято с http://evolution.berkeley.edu/evolibrary/article/side_0_0/evo_06
Видео ниже фокусируется на терминологии и исследует некоторые неправильные представления о чтении деревьев:
 youtube.com/embed/RVW8iIEfiZI?feature=oembed» frameborder=»0″ allow=»accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture» allowfullscreen=»»>
youtube.com/embed/RVW8iIEfiZI?feature=oembed» frameborder=»0″ allow=»accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture» allowfullscreen=»»>
Неправильные представления и как правильно читать филогенетическое дерево
Деревья могут сбивать с толку. Распространенная ошибка — читать верхушки деревьев и думать, что их порядок имеет значение. В приведенном выше дереве ближайший родственник таксона C не является таксоном B. И A, и B одинаково удалены от таксона C или связаны с ним. Фактически, перестановка меток таксонов A и B приведет к топологически эквивалентному дереву. . Важен порядок ветвления по оси времени. На приведенном ниже рисунке показано, что можно вращать ветки, не затрагивая структуру дерева, подобно подвесному мобилю:
http://evolution.berkeley.edu/evolibrary/article/%3C?%20echo%20$baseURL;%20?%3E_0_0/evotrees_primer_08
Висячий мобиль для птиц от Чарли Харпера
Также может быть трудно распознать как деревья моделируют эволюционные отношения. Следует помнить, что любое дерево представляет собой ничтожную часть дерева жизни.
Следует помнить, что любое дерево представляет собой ничтожную часть дерева жизни.
Учитывая только дерево из 5 таксонов (без пунктирных ветвей), заманчиво думать, что таксон S является наиболее «примитивным» или наиболее похожим на общего предка, представленного корневым узлом, поскольку между ними нет дополнительных узлов. С и корень. Однако в ходе эволюции, несомненно, было много ответвлений от этой линии, большинство из которых привело к вымершим таксонам (9).9% всех видов считаются вымершими), а многие из них относятся к живым таксонам (например, фиолетовая пунктирная линия), которые просто не показаны на дереве. В таком случае имеет значение общее расстояние по оси времени (вертикальная ось в этом дереве) — таксон S эволюционировал в течение 5 миллионов лет, то есть столько же времени, сколько и любой из других 4 таксонов. Поскольку дерево нарисовано с вертикальной осью времени, горизонтальная ось не имеет значения и служит только для разделения таксонов и их родословных. Таким образом, ни один из ныне живущих таксонов не является ни более «примитивным», ни более «продвинутым», чем любой другой; все они эволюционировали в течение одинакового периода времени от своего последнего общего предка.
Таким образом, ни один из ныне живущих таксонов не является ни более «примитивным», ни более «продвинутым», чем любой другой; все они эволюционировали в течение одинакового периода времени от своего последнего общего предка.
Ось времени также позволяет нам количественно измерять эволюционные расстояния. Расстояние между A и Q составляет 4 миллиона лет (A эволюционировала в течение 2 миллионов лет с момента их разделения, а Q также развивалась независимо от A в течение 2 миллионов лет после разделения). Расстояние между A и D составляет 6 миллионов лет, так как они отделились от своего общего предка 3 миллиона лет назад.
Филогенетические деревья могут иметь разную форму – они могут быть ориентированы боком, перевернуты (самые свежие внизу), ветви могут быть изогнутыми, или дерево может быть радиальным (самые старые в центре). Независимо от того, как нарисовано дерево, все модели ветвления передают одну и ту же информацию: эволюционное происхождение и модели дивергенции.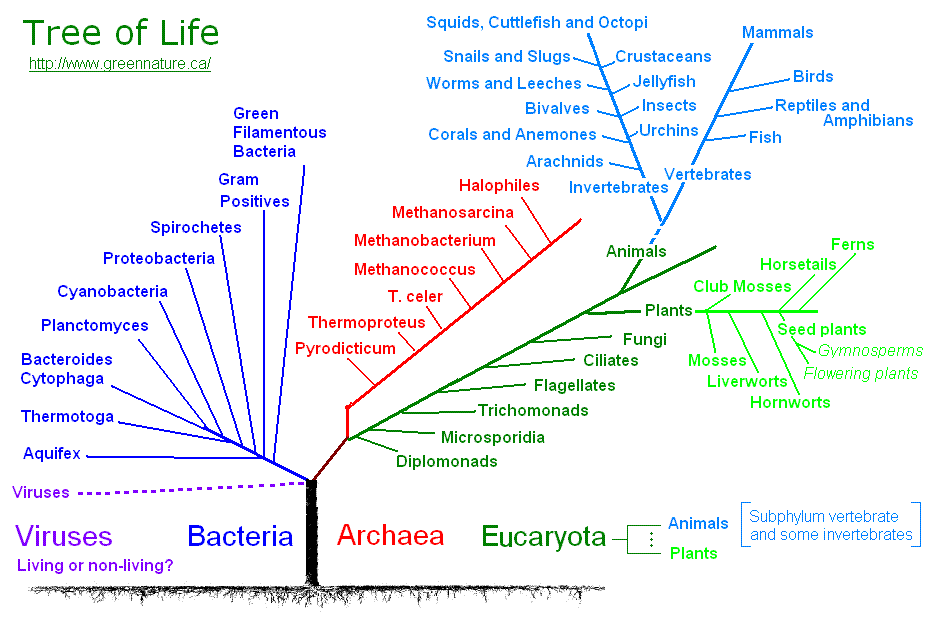
Это видео отлично объясняет, как интерпретировать родство видов с помощью деревьев, включая описание некоторых неправильных способов чтения деревьев:
Построение филогенетических деревьев
Для построить филогенетические деревья, включая морфологические данные, такие как структурные особенности, типы органов и специфические скелетные устройства; и генетические данные, такие как последовательности митохондриальной ДНК, гены рибосомной РНК и любые представляющие интерес гены.
Эти типы данных используются для определения гомологии, что означает сходство из-за общего происхождения. Это просто идея о том, что вы наследуете черты от своих родителей, только применяемая на уровне вида: у всех людей большой мозг и противопоставленные большие пальцы, потому что так было у наших предков; все млекопитающие производят молоко из молочных желез, потому что это делали их предки.
Деревья строятся по принципу экономичности, который заключается в том, что наиболее вероятным шаблоном является тот, который требует наименьшего количества изменений. Например, гораздо более вероятно, что все млекопитающие производят молоко, потому что все они унаследовали молочные железы от общего предка, который производил молоко из молочных желез, по сравнению с несколькими группами организмов, каждая из которых развивала молочные железы независимо.
Вот отличный ресурс по филогенетическим деревьям: https://evolution.berkeley.edu/evolibrary/article/0_0_0/evotrees_intro
Филогенетические деревья | Биология для специальностей I
Прочитайте и проанализируйте филогенетическое дерево, которое документирует эволюционные отношения
В научных терминах история эволюции и взаимоотношения организма или группы организмов называется филогенезом. Филогения описывает отношения организма, например, от каких организмов он, как считается, произошел, с какими видами он наиболее тесно связан и так далее. Филогенетические отношения предоставляют информацию об общем происхождении, но не обязательно о том, чем организмы похожи или отличаются.
Филогенетические отношения предоставляют информацию об общем происхождении, но не обязательно о том, чем организмы похожи или отличаются.
Цели обучения
- Выяснить, как и почему ученые классифицируют организмы на Земле
- Различать типы филогенетических деревьев и то, что говорит нам их структура
- Определите некоторые ограничения филогенетических деревьев
- Связь системы таксономической классификации и биномиальной номенклатуры
Научная классификация
Рисунок 1. В этой коллекции жуков представлены лишь некоторые из более чем одного миллиона известных видов насекомых. Жуки — крупная подгруппа насекомых. Они составляют около 40 процентов всех видов насекомых и около 25 процентов всех известных видов организмов.
Почему биологи классифицируют организмы? Основная причина заключается в том, чтобы понять невероятное разнообразие жизни на Земле. Ученые идентифицировали миллионы различных видов организмов. Среди животных наиболее разнообразной группой организмов являются насекомые. Описано более миллиона различных видов насекомых. По оценкам, девять миллионов видов насекомых еще предстоит идентифицировать. Крошечная часть видов насекомых показана в коллекции жуков на рисунке 1.
Описано более миллиона различных видов насекомых. По оценкам, девять миллионов видов насекомых еще предстоит идентифицировать. Крошечная часть видов насекомых показана в коллекции жуков на рисунке 1.
Какими бы разнообразными ни были насекомые, видов бактерий, еще одной крупной группы организмов, может быть еще больше. Ясно, что существует необходимость организовать огромное многообразие жизни. Классификация позволяет ученым организовать и лучше понять основные сходства и различия между организмами. Эти знания необходимы для понимания настоящего разнообразия и прошлой эволюционной истории жизни на Земле.
Филогенетические деревья
Ученые используют инструмент, называемый филогенетическим деревом, чтобы показать эволюционные пути и связи между организмами. А Филогенетическое дерево — это диаграмма, используемая для отражения эволюционных отношений между организмами или группами организмов. Ученые считают филогенетические деревья гипотезой эволюционного прошлого, поскольку невозможно вернуться назад, чтобы подтвердить предполагаемые отношения. Другими словами, можно построить «древо жизни», чтобы проиллюстрировать эволюцию различных организмов и показать взаимосвязь между разными организмами (рис. 2).
Другими словами, можно построить «древо жизни», чтобы проиллюстрировать эволюцию различных организмов и показать взаимосвязь между разными организмами (рис. 2).
Каждая группа организмов прошла свой эволюционный путь, называемый филогенезом. Каждый организм связан родством с другими, и, основываясь на морфологических и генетических данных, ученые пытаются составить карту эволюционных путей всей жизни на Земле. Многие ученые строят филогенетические деревья, чтобы проиллюстрировать эволюционные отношения.
Структура филогенетических деревьев
Филогенетическое древо можно читать как карту истории эволюции. Многие филогенетические деревья имеют в основе одну линию, представляющую общего предка. Ученые называют такие деревья корневыми, что означает наличие единой линии предков (обычно нарисованной снизу или слева), к которой относятся все организмы, представленные на диаграмме. Обратите внимание, что на корневом филогенетическом дереве три домена — бактерии, археи и эукариоты — расходятся из одной точки и ответвляются. Небольшая ветвь, занимаемая растениями и животными (включая человека) на этой диаграмме, показывает, насколько недавно и ничтожны эти группы по сравнению с другими организмами. Неукорененные деревья не показывают общего предка, но показывают отношения между видами.
Небольшая ветвь, занимаемая растениями и животными (включая человека) на этой диаграмме, показывает, насколько недавно и ничтожны эти группы по сравнению с другими организмами. Неукорененные деревья не показывают общего предка, но показывают отношения между видами.
Рисунок 2. Оба этих филогенетических дерева показывают взаимосвязь между тремя доменами жизни — бактериями, археями и эукариями — но (а) корневое дерево пытается определить, когда различные виды отделились от общего предка, в то время как (б) неукорененное дерево — нет. (кредит а: модификация работы Эрика Габа)
В корневом дереве ветвление указывает на эволюционные отношения (рис. 3). Точка, в которой происходит разделение, называемая точкой ветвления , представляет собой место, где одна линия развилась в отдельную новую. Линия, которая рано развилась от корня и остается неразветвленной, называется 9.0023 базальный таксон . Когда две линии происходят от одной и той же точки ветвления, их называют сестринскими таксонами . Ветвь с более чем двумя родословными называется политомией и служит иллюстрацией того, где ученые не определили окончательно все отношения. Важно отметить, что, хотя сестринские таксоны и политомии имеют общего предка, это не означает, что группы организмов отделились или произошли друг от друга. Организмы двух таксонов могли разделиться в определенной точке ветвления, но ни один таксон не дал начало другому.
Ветвь с более чем двумя родословными называется политомией и служит иллюстрацией того, где ученые не определили окончательно все отношения. Важно отметить, что, хотя сестринские таксоны и политомии имеют общего предка, это не означает, что группы организмов отделились или произошли друг от друга. Организмы двух таксонов могли разделиться в определенной точке ветвления, но ни один таксон не дал начало другому.
Рисунок 3. Корень филогенетического дерева указывает на то, что родовая линия дала начало всем организмам на дереве. Точка ветвления указывает, где разошлись две родословные. Линия, которая развилась рано и остается неразветвленной, является базальным таксоном. Когда две линии происходят из одной и той же точки ветвления, они являются сестринскими таксонами. Ветвь с более чем двумя родословными является политомией.
Диаграммы выше могут служить путем к пониманию истории эволюции. Путь можно проследить от зарождения жизни до любого отдельного вида, перемещаясь по эволюционным ветвям между двумя точками. Кроме того, начав с одного вида и проследив обратно к «стволу» дерева, можно обнаружить предков этого вида, а также узнать, где родословные имеют общее происхождение. Кроме того, дерево можно использовать для изучения целых групп организмов.
Кроме того, начав с одного вида и проследив обратно к «стволу» дерева, можно обнаружить предков этого вида, а также узнать, где родословные имеют общее происхождение. Кроме того, дерево можно использовать для изучения целых групп организмов.
Еще один момент, который следует упомянуть о структуре филогенетического дерева, заключается в том, что вращение в точках ветвления не меняет информацию. Например, если повернуть точку ветвления и изменить порядок таксонов, это не изменит информацию, потому что эволюция каждого таксона из точки ветвления не зависит от другого.
Многие дисциплины в рамках изучения биологии способствуют пониманию того, как жизнь в прошлом и настоящем развивалась с течением времени; вместе эти дисциплины способствуют построению, обновлению и поддержанию «дерева жизни». Информация используется для организации и классификации организмов на основе эволюционных отношений в научной области, называемой систематикой. Данные могут быть собраны из окаменелостей, путем изучения структуры частей тела или молекул, используемых организмом, а также с помощью анализа ДНК.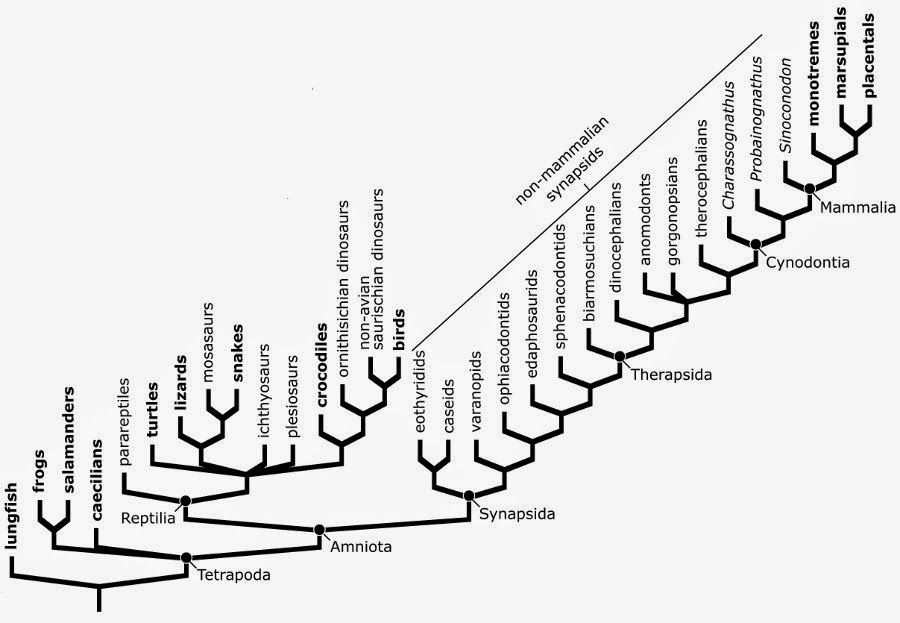 Комбинируя данные из многих источников, ученые могут составить филогению организма; поскольку филогенетические деревья являются гипотезами, они будут продолжать меняться по мере открытия новых типов жизни и получения новой информации.
Комбинируя данные из многих источников, ученые могут составить филогению организма; поскольку филогенетические деревья являются гипотезами, они будут продолжать меняться по мере открытия новых типов жизни и получения новой информации.
Видеообзор
Ограничения филогенетических деревьев
Легко предположить, что более близкородственные организмы выглядят более похожими, и хотя это часто бывает, это не всегда так. Если две близкородственные линии развились в значительно различающихся условиях или после эволюции крупной новой адаптации, две группы могут казаться более разными, чем другие группы, которые не так тесно связаны. Например, филогенетическое дерево на рисунке 4 показывает, что и у ящериц, и у кроликов есть амниотические яйца, а у лягушек их нет; однако ящерицы и лягушки кажутся более похожими, чем ящерицы и кролики.
Рисунок 4. Это лестничное филогенетическое древо позвоночных основано на организме, у которого отсутствует позвоночник.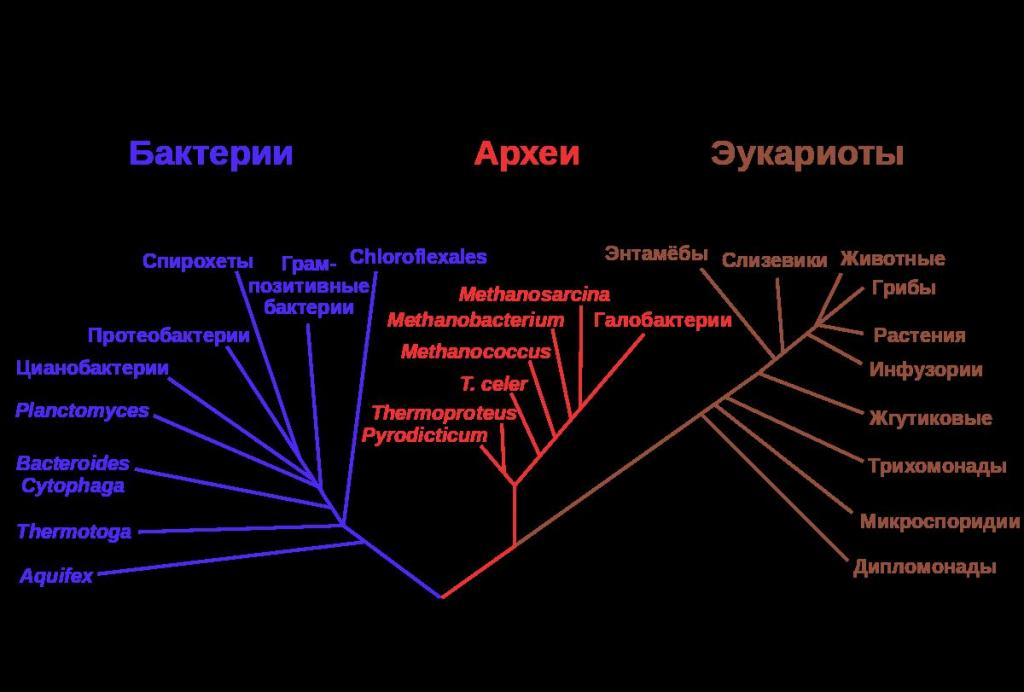 В каждой точке ветвления организмы с разными признаками помещаются в разные группы в зависимости от общих характеристик.
В каждой точке ветвления организмы с разными признаками помещаются в разные группы в зависимости от общих характеристик.
Другой аспект филогенетических деревьев заключается в том, что, если не указано иное, ветви не учитывают продолжительность времени, а только эволюционный порядок. Другими словами, длина ветви обычно не означает, что прошло больше времени, а короткая ветвь не означает, что прошло меньше времени, если только это не указано на диаграмме. Например, на рисунке 4 дерево не показывает, сколько времени прошло между эволюцией амниотических яиц и волос. То, что показывает дерево, — это порядок, в котором происходили события. Снова используя рисунок 4, дерево показывает, что самым старым признаком является позвоночный столб, за которым следуют шарнирные челюсти и так далее. Помните, что любое филогенетическое дерево является частью большего целого, и, подобно настоящему дереву, оно не растет только в одном направлении после развития новой ветви.
Таким образом, для организмов на рис. 4 то, что развился позвоночник, не означает, что эволюция беспозвоночных прекратилась, это означает лишь то, что образовалась новая ветвь. Кроме того, группы, которые не являются близкородственными, но развиваются в сходных условиях, могут казаться фенотипически более похожими друг на друга, чем на близкого родственника.
4 то, что развился позвоночник, не означает, что эволюция беспозвоночных прекратилась, это означает лишь то, что образовалась новая ветвь. Кроме того, группы, которые не являются близкородственными, но развиваются в сходных условиях, могут казаться фенотипически более похожими друг на друга, чем на близкого родственника.
Посетите этот веб-сайт, чтобы увидеть интерактивные упражнения, которые позволят вам исследовать эволюционные отношения между видами.
Система таксономической классификации
Таксономия (что буквально означает «закон упорядочения») — это наука о классификации организмов для создания общедоступных на международном уровне систем классификации, в которых каждый организм помещается во все более и более всеобъемлющие группы. Подумайте о том, как организован продуктовый магазин. Одно большое пространство разделено на отделы, такие как продуктовый, молочный и мясной. Затем каждый отдел делится на проходы, затем каждый проход делится на категории и бренды, а затем, наконец, один продукт.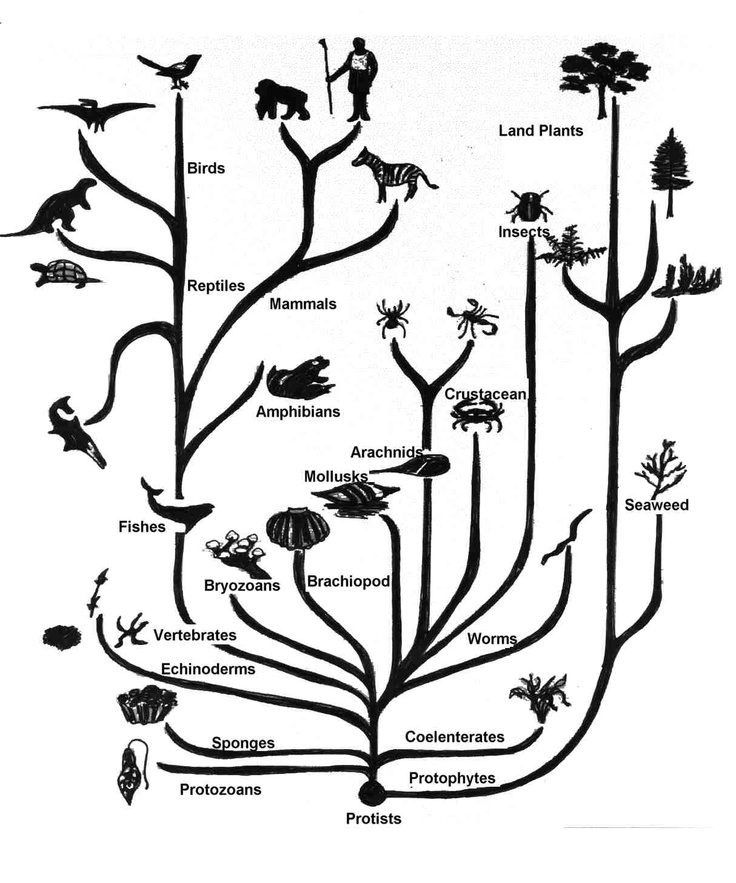 Эта организация от больших к меньшим, более конкретным категориям называется иерархической системой.
Эта организация от больших к меньшим, более конкретным категориям называется иерархической системой.
Система таксономической классификации (также называемая системой Линнея по имени ее изобретателя Карла Линнея, шведского ботаника, зоолога и врача) использует иерархическую модель. По мере продвижения от места возникновения группы становятся более специфичными, пока одна ветвь не заканчивается единым видом. Например, после общего начала всей жизни ученые делят организмы на три большие категории, называемые доменом: бактерии, археи и эукариоты. В каждом домене есть вторая категория, называемая королевство . После царств следуют категории возрастающей специфичности: тип , класс , порядок , семейство , род и вид (рис. 5).
Рисунок 5. Система таксономической классификации использует иерархическую модель для организации живых организмов во все более конкретные категории.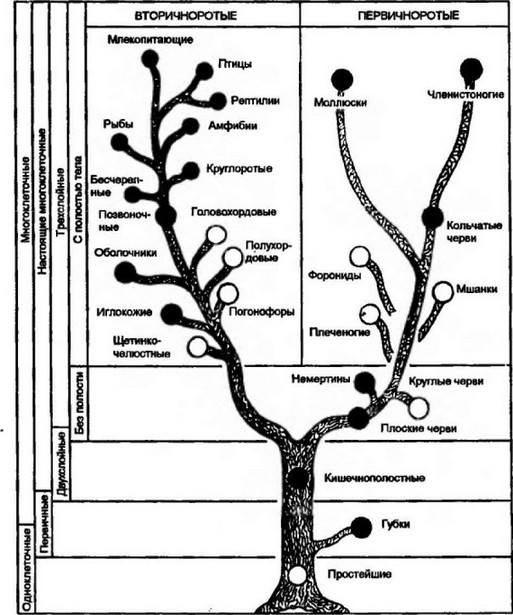 Обыкновенная собака, Canis lupus familiaris , является подвидом Canis lupus , который также включает волка и динго. (кредит «собака»: модификация работы Janneke Vreugdenhil)
Обыкновенная собака, Canis lupus familiaris , является подвидом Canis lupus , который также включает волка и динго. (кредит «собака»: модификация работы Janneke Vreugdenhil)
Царство Animalia происходит от домена Eukarya. Для обычной собаки уровни классификации будут такими, как показано на рисунке 5. Таким образом, полное название организма технически состоит из восьми терминов. Для собак это: Eukarya, Animalia, Chordata, Mammalia, Carnivora, Canidae, Canis, и lupus . Обратите внимание, что все названия пишутся с заглавной буквы, кроме видов, а названия родов и видов выделены курсивом. Ученые обычно ссылаются на организм только по его роду и виду, что является его научным названием, состоящим из двух слов, в том, что называется биномиальная номенклатура . Поэтому научное название собаки Canis lupus . Название на каждом уровне также называется таксоном . Другими словами, собаки в порядке плотоядные.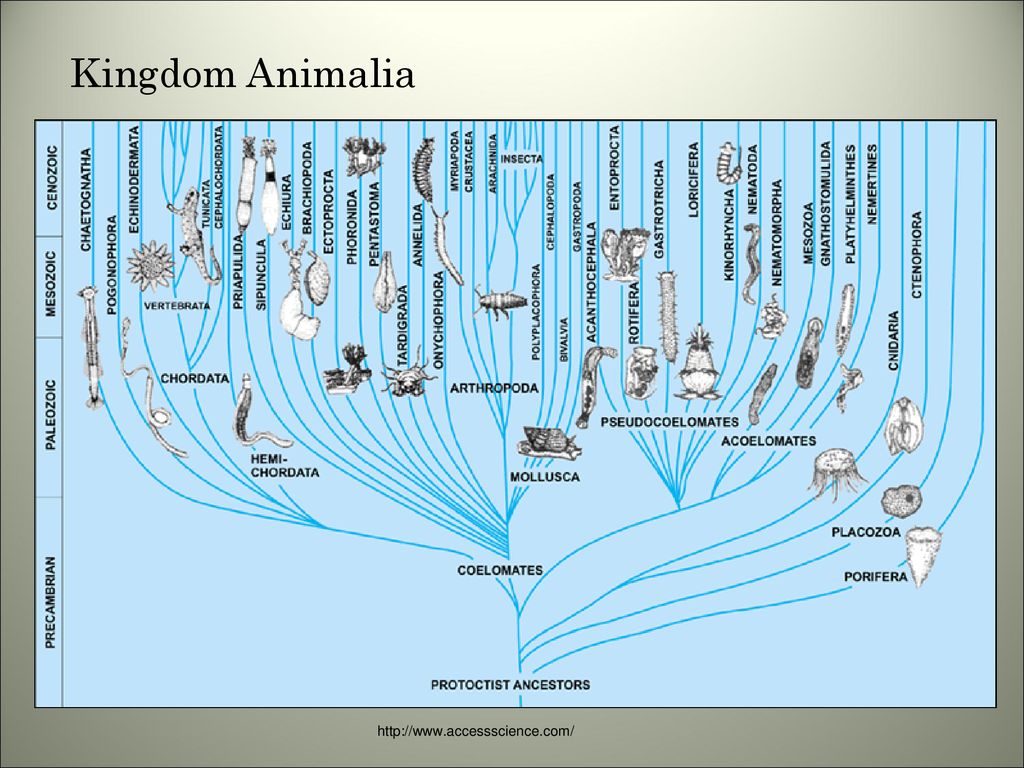 Carnivora — название таксона на уровне отряда; Canidae — таксон на уровне семейства и так далее. Организмы также имеют общее название, которое люди обычно используют, в данном случае собака. Обратите внимание, что собака также является подвидом: « Familiaris » в Canis lupus Familiis. Подвиды — это представители одного и того же вида, которые способны спариваться и воспроизводить жизнеспособное потомство, но считаются отдельными подвидами из-за географической или поведенческой изоляции или других факторов.
Carnivora — название таксона на уровне отряда; Canidae — таксон на уровне семейства и так далее. Организмы также имеют общее название, которое люди обычно используют, в данном случае собака. Обратите внимание, что собака также является подвидом: « Familiaris » в Canis lupus Familiis. Подвиды — это представители одного и того же вида, которые способны спариваться и воспроизводить жизнеспособное потомство, но считаются отдельными подвидами из-за географической или поведенческой изоляции или других факторов.
На рисунке 6 показано, как уровни приближаются к специфичности для других организмов. Обратите внимание, что собака делит территорию с самыми разнообразными организмами, включая растения и бабочек. На каждом подуровне организмы становятся более похожими, потому что они более тесно связаны. Исторически сложилось так, что ученые классифицировали организмы по признакам, но по мере развития технологии ДНК были определены более точные филогении.
Практический вопрос
Рисунок 6. На каждом подуровне системы таксономической классификации организмы становятся более похожими. Собаки и волки — это один и тот же вид, потому что они могут размножаться и производить жизнеспособное потомство, но они достаточно разные, чтобы их можно было отнести к разным подвидам. (кредит «растение»: модификация работы «berduchwal»/Flickr; кредит «насекомое»: модификация работы Джона Салливана; кредит «рыба»: модификация работы Кристиана Мелфюрера; кредит «кролик»: модификация работы Эйдана Войтас; кредит «кошка»: модификация работы Джонатана Лидбека; кредит «лиса»: модификация работы Кевина Бахера, NPS; кредит «шакал»: модификация работы Томаса А. Херманна, NBII, Геологическая служба США; кредит «волк» : модификация работы Роберта Дьюара; кредит «собака»: модификация работы «digital_image_fan»/Flickr)
На каждом подуровне системы таксономической классификации организмы становятся более похожими. Собаки и волки — это один и тот же вид, потому что они могут размножаться и производить жизнеспособное потомство, но они достаточно разные, чтобы их можно было отнести к разным подвидам. (кредит «растение»: модификация работы «berduchwal»/Flickr; кредит «насекомое»: модификация работы Джона Салливана; кредит «рыба»: модификация работы Кристиана Мелфюрера; кредит «кролик»: модификация работы Эйдана Войтас; кредит «кошка»: модификация работы Джонатана Лидбека; кредит «лиса»: модификация работы Кевина Бахера, NPS; кредит «шакал»: модификация работы Томаса А. Херманна, NBII, Геологическая служба США; кредит «волк» : модификация работы Роберта Дьюара; кредит «собака»: модификация работы «digital_image_fan»/Flickr)
На каком уровне кошки и собаки считаются частью одной группы?
Показать ответ
Посетите этот веб-сайт, чтобы классифицировать три организма — медведя, орхидею и морской огурец — от царства к виду. Чтобы запустить игру, в разделе «Классификация жизни» щелкните изображение медведя или кнопку «Запустить интерактив».
Чтобы запустить игру, в разделе «Классификация жизни» щелкните изображение медведя или кнопку «Запустить интерактив».
Недавний генетический анализ и другие достижения показали, что некоторые более ранние филогенетические классификации не соответствуют эволюционному прошлому; поэтому изменения и обновления необходимо вносить по мере появления новых открытий. Напомним, что филогенетические деревья являются гипотезами и модифицируются по мере поступления данных. Кроме того, классификация исторически была сосредоточена на группировке организмов в основном по общим характеристикам и не обязательно иллюстрировала, как различные группы соотносятся друг с другом с эволюционной точки зрения. Например, несмотря на то, что гиппопотам больше похож на свинью, чем на кита, гиппопотам может быть ближайшим живым родственником кита.
Проверьте свое понимание
Ответьте на вопросы ниже, чтобы узнать, насколько хорошо вы понимаете темы, затронутые в предыдущем разделе. В этом коротком тесте , а не учитываются при подсчете вашей оценки в классе, и вы можете пересдавать его неограниченное количество раз.
В этом коротком тесте , а не учитываются при подсчете вашей оценки в классе, и вы можете пересдавать его неограниченное количество раз.
Используйте этот тест, чтобы проверить свое понимание и решить, следует ли (1) изучить предыдущий раздел дальше или (2) перейти к следующему разделу.
Вывод филогенетического дерева: нисходящий подход к отслеживанию эволюции опухоли
Введение
Рак — это эволюционный процесс, который формируется под давлением отбора и накоплением соматических мутаций, что приводит к высокому уровню гетерогенности внутри и между образцами опухолей (Marusyk et al., 2012; Yates and Campbell, 2012). Такая неоднородность геномов может быть использована для выделения субклональных популяций опухолей и отслеживания эволюционной траектории развития рака. Метастазирование обычно считается последней стадией прогрессирования рака и по-прежнему является основной причиной смерти от рака, но его механизм плохо изучен. В ряде исследований секвенировали множественные биопсии первичных и метастатических опухолей, чтобы выяснить порядок накопления мутаций и происхождение дистальных метастазов (Gundem et al.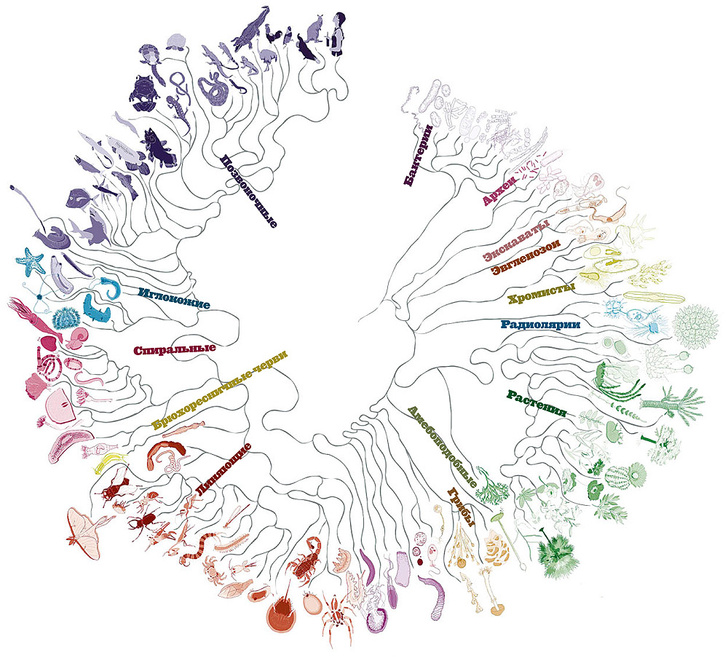 , 2015; Yates et al., 2017; Ferronika et al., 2019).). Лучшее понимание процесса метастазирования может в конечном итоге привести к новым стратегиям диагностики и лечения.
, 2015; Yates et al., 2017; Ferronika et al., 2019).). Лучшее понимание процесса метастазирования может в конечном итоге привести к новым стратегиям диагностики и лечения.
Существует ряд вычислительных методов для определения генотипов популяций опухолевых клеток. Однако большинство существующих методов делают вывод о филогении эволюции рака на основе частот соматических мутаций вариантных аллелей (VAF) из данных глубокого секвенирования ДНК (Jiao et al., 2014; Malikic et al., 2015; Yates et al., 2015; Nieboer et al. др., 2018). В нескольких традиционных методах филогенетического вывода используется множественное выравнивание последовательностей, объединение соседей с корреляционными расстояниями Пирсона, алгоритм максимальной экономии или алгоритм максимального правдоподобия, основанный на наличии вариантов в образцах (Kim et al., 2015; Lu et al., 2016; Zhao et al. , 2016; Чой и др., 2017; Наксерова и др., 2017; Чжай и др., 2017). Большинство из этих методов требуют больших вычислительных ресурсов и длительного времени выполнения.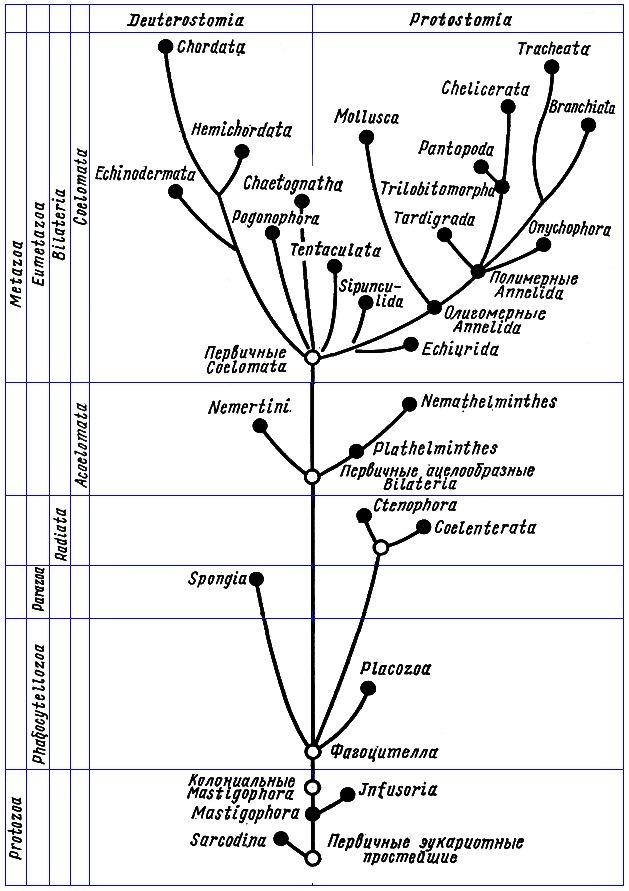 В 2015 году был разработан LICHeE для построения филогенетических деревьев опухолей с несколькими образцами и субклональной декомпозиции опухолей из точных VAF соматических однонуклеотидных вариантов (SSNV), полученных путем глубокого секвенирования (Popic et al., 2015). LICHeE сначала группирует подмножества соматических мутаций, которые имеют сходные паттерны присутствия-отсутствия, а также аналогичные VAF в нескольких образцах опухолей. Затем он строит ограниченную сеть, чтобы вывести отношения между кластерами соматических мутаций и идентифицировать филогенетические деревья опухолей. Несколько других методов используют аналогичные принципы, но другие методологические основы, такие как Treeomics и BAMSE (Reiter et al., 2017; Toosi et al., 2019).). Триомика была разработана для реконструкции филогении метастазов и сопоставления субклонов с их анатомическим расположением. В качестве входных файлов он использует общее количество прочтений и вариантных прочтений SSNV из нескольких связанных нормальных и опухолевых образцов отдельных больных раком.
В 2015 году был разработан LICHeE для построения филогенетических деревьев опухолей с несколькими образцами и субклональной декомпозиции опухолей из точных VAF соматических однонуклеотидных вариантов (SSNV), полученных путем глубокого секвенирования (Popic et al., 2015). LICHeE сначала группирует подмножества соматических мутаций, которые имеют сходные паттерны присутствия-отсутствия, а также аналогичные VAF в нескольких образцах опухолей. Затем он строит ограниченную сеть, чтобы вывести отношения между кластерами соматических мутаций и идентифицировать филогенетические деревья опухолей. Несколько других методов используют аналогичные принципы, но другие методологические основы, такие как Treeomics и BAMSE (Reiter et al., 2017; Toosi et al., 2019).). Триомика была разработана для реконструкции филогении метастазов и сопоставления субклонов с их анатомическим расположением. В качестве входных файлов он использует общее количество прочтений и вариантных прочтений SSNV из нескольких связанных нормальных и опухолевых образцов отдельных больных раком.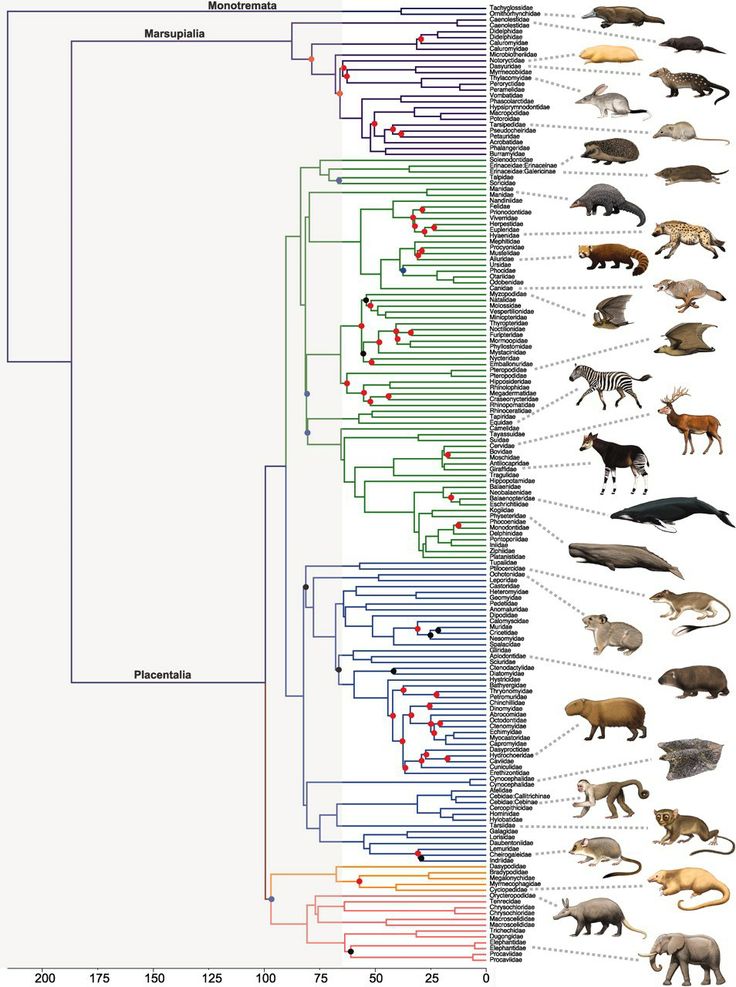 Затем он использует байесовскую модель вывода для выявления эволюционно совместимых моделей мутаций, а затем выводит эволюционные деревья. Другой вероятностный метод, названный BAMSE, делает вывод о субклональной истории и реконструкции дерева генезиса гетерогенных образцов опухолей, используя в качестве входных данных количество прочтений соматических мутаций. Апостериорная вероятность дерева выводится с помощью байесовской модели, которая объединяет априорные представления о количестве субклонов, составе опухоли и процессе субклональной эволюции. Однако пользователи должны решить, сколько субклонов, что обычно трудно оценить. Есть две основные проблемы, общие для этих методов. Что наиболее важно, часто бывает трудно получить точную частоту аллеля из клинических образцов, таких как образцы, фиксированные формалином и залитые парафином (FFPE) (Astolfi et al., 2015). Результаты этих методов также чувствительны к нескольким ключевым параметрам, и тем не менее у пользователей нет простого способа выбрать эти параметры.
Затем он использует байесовскую модель вывода для выявления эволюционно совместимых моделей мутаций, а затем выводит эволюционные деревья. Другой вероятностный метод, названный BAMSE, делает вывод о субклональной истории и реконструкции дерева генезиса гетерогенных образцов опухолей, используя в качестве входных данных количество прочтений соматических мутаций. Апостериорная вероятность дерева выводится с помощью байесовской модели, которая объединяет априорные представления о количестве субклонов, составе опухоли и процессе субклональной эволюции. Однако пользователи должны решить, сколько субклонов, что обычно трудно оценить. Есть две основные проблемы, общие для этих методов. Что наиболее важно, часто бывает трудно получить точную частоту аллеля из клинических образцов, таких как образцы, фиксированные формалином и залитые парафином (FFPE) (Astolfi et al., 2015). Результаты этих методов также чувствительны к нескольким ключевым параметрам, и тем не менее у пользователей нет простого способа выбрать эти параметры.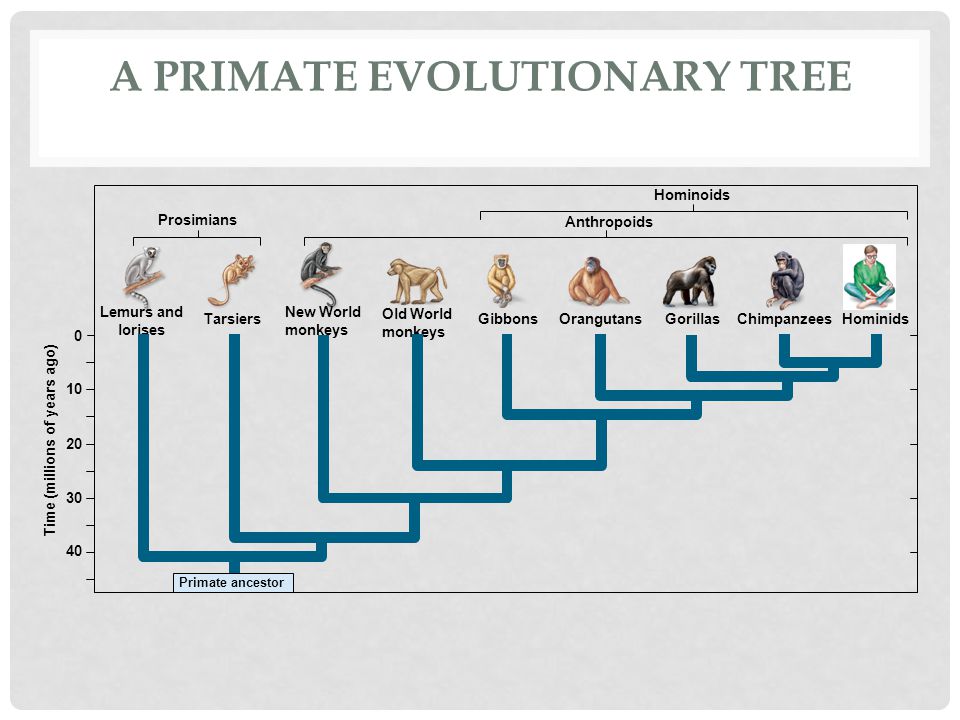
Здесь мы предлагаем PTI (вывод филогенетического дерева), новый метод, который использует итеративный нисходящий подход для вывода филогенетической древовидной структуры нескольких биоптатов опухоли одного и того же пациента с использованием соматических мутаций без необходимости точного определения частот аллелей. Кроме того, у PTI есть только один параметр для установки, и мы также предоставляем четкие инструкции о том, как установить этот параметр.
Методы
PTI — это метод, разработанный для использования итеративного нисходящего подхода для построения корневого филогенетического дерева среди нескольких образцов одного и того же пациента. В этом разделе мы представляем обзор нашего подхода (рис. 1). Во-первых, PTI идентифицирует общие мутации для всех образцов и определяет количество общих мутаций как длину корневого ствола. Затем PTI использует итеративный нисходящий подход, чтобы найти оптимальное разделение ветвей, пока все выборки не достигнут конечных узлов. PTI также аннотирует мутации известных драйверных генов в древовидной структуре, что облегчает интуитивное понимание ключевых событий мутации во время прогрессирования рака.
PTI также аннотирует мутации известных драйверных генов в древовидной структуре, что облегчает интуитивное понимание ключевых событий мутации во время прогрессирования рака.
Рисунок 1 Обзор PTI. (A) Рабочий процесс PTI. (B) На примере пациента с шестью образцами подробно показан специфический процесс PTI для вывода филогенетического дерева. Квадратная сетка указывает на то, что конечный узел был достигнут, а кружок указывает на то, что оставшиеся выборки все еще необходимо повторить, чтобы найти наилучшее разделение ветвей. (C) Пример филогенетического дерева, полученного с помощью PTI. Филогенетическое дерево — это корневое дерево, листовые узлы которого являются образцами. Информация аннотации ветвей включает в себя длину ветвей, которая равна количеству общих мутаций, и аннотацию генов-драйверов.
Идентификация и удаление общих мутаций из всех образцов
Предположим, мы получили несколько образцов s , s ∈ {1,2,…, n } от одного и того же пациента. Используя вызывающие соматические мутации, такие как Mutect2 или VarScan2 (Koboldt et al., 2012; Cibulskis et al., 2013), мы можем определить частоты мутационных аллелей в каждом образце. Определите эти соматические мутации как r 1 , r 2 …, r m . Затем мы строим бинарную матрицу M с рядами с надписью R 1 , R 2 …, R M и колонны, помеченные S и колонны, помеченные S и колонны и колонны и колонны. , так что M ij = 1 тогда и только тогда, когда VAF соматической мутации r i в образце s j больше или равен заданному порогу. Чем больше мутаций с высокой степенью достоверности используется для построения филогенетического дерева, тем точнее структура дерева. Однако, когда количество соматических мутаций у пациента слишком велико, PTI может реализовать необязательный этап фильтрации соматических мутаций на основе частоты аллеля (по умолчанию 0,1).
Используя вызывающие соматические мутации, такие как Mutect2 или VarScan2 (Koboldt et al., 2012; Cibulskis et al., 2013), мы можем определить частоты мутационных аллелей в каждом образце. Определите эти соматические мутации как r 1 , r 2 …, r m . Затем мы строим бинарную матрицу M с рядами с надписью R 1 , R 2 …, R M и колонны, помеченные S и колонны, помеченные S и колонны и колонны и колонны. , так что M ij = 1 тогда и только тогда, когда VAF соматической мутации r i в образце s j больше или равен заданному порогу. Чем больше мутаций с высокой степенью достоверности используется для построения филогенетического дерева, тем точнее структура дерева. Однако, когда количество соматических мутаций у пациента слишком велико, PTI может реализовать необязательный этап фильтрации соматических мутаций на основе частоты аллеля (по умолчанию 0,1). Матрица M — вход PTI. Мы вычисляем пересечение мутаций во всех выборках одного и того же пациента и определяем пересечение как длину корневого ствола. После удаления общих мутаций из всех образцов следующим шагом является поиск оптимального разделения ветвей для отфильтрованного набора данных M filter .
Матрица M — вход PTI. Мы вычисляем пересечение мутаций во всех выборках одного и того же пациента и определяем пересечение как длину корневого ствола. После удаления общих мутаций из всех образцов следующим шагом является поиск оптимального разделения ветвей для отфильтрованного набора данных M filter .
Найдите оптимальное разделение ветвей
Геномные вариации в раковых клетках постепенно накапливаются в ходе канцерогенеза и развития рака (Gerlinger et al., 2012; Sato et al., 2016). Таким образом, несмотря на сложность эволюции рака, в существующих исследованиях на эволюционном дереве редко наблюдается разделение более чем на два пути (Hong et al., 2015; Schwarz et al., 2015; Brown et al., 2017). Следовательно, PTI будет перебирать все возможные двусторонние разделения ветвей, S={(1,n−1),(2,n−2),…(|n2|,n−|n2|)}, чтобы вывести оптимальное раскол ветки. Примечательно, что наш метод действительно способен обнаруживать более двух способов разделения в любом заданном эволюционном узле (дополнительное примечание 1).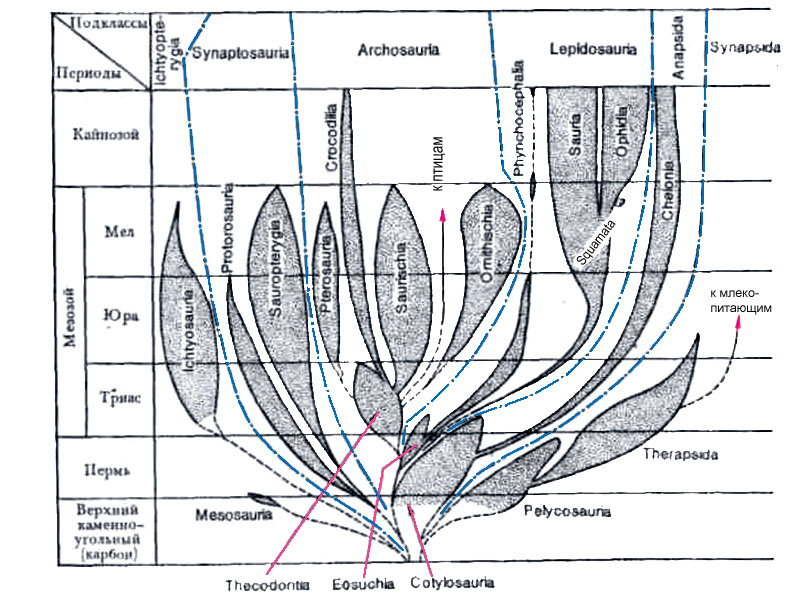
Для каждого возможного отделения ответвления S t ,
Включено t∈{1,2,…,|n2|} ,∁nt комбинаций. Пусть θ будет объектом чисел общих мутаций, измеренных на всех возможных расщеплениях ветвей. Для каждого возможного разделения ветвей S t и комбинации c , c∈{1,2,…,∁nt} соответствующий элемент θ tc представляет количество общих мутаций в большей группе. Если n четно, нет большей группы возможного расщепления ветвей (n2,n−n2). Тогда в этом случае θn2c представляет меньшее количество общих мутаций в двух группах одинакового размера.
Чтобы определить, какое возможное разделение ветвей является лучшим, мы определяем ∂ как вектор отношения, измеренный для всех возможных разделений ветвей, который рассчитывается по уравнению (1), где θ t max представляет Максимальное значение и θ T SEC_MAX представляет вторичное максимальное значение в θ T :
2 ∂T = θt Maxθt SEC_MAX (1)
∂T = θt Maxθt SEC_MAX (1)
∂T = θt Maxθt SEC_MAX (1)
∂T = θt maxθt sec_max (1)
∂t = θt maxθt sec_max (1)
∂t = θt maxθt sec_max (1)
∂t = θ0276 , то соотношение между лучшей комбинацией и второй лучшей комбинацией должно быть намного больше по сравнению с неоптимальными разбиениями (подробное описание обоснования использования этого соотношения см. в дополнительном примечании 2).
в дополнительном примечании 2).
Затем выборки, достигшие конечного узла после оптимального разделения, будут удалены из набора данных M filter . Этот метод будет перебирать остальные образцы, пока все образцы не будут разделены на конечные узлы. Для пациента может существовать более одной древовидной структуры с одинаковым значением ∂. Чтобы определить оптимальную древовидную структуру, агрегированный подсчет мутаций ( W T ) вычисляется для каждой древовидной структуры с использованием мутаций на всех стволах, содержащих два или более листовых узла. Дерево с наибольшим количеством баллов будет оптимальным деревом. Пусть i , i ∈ {0,1,…, k } представляет все уровни ствола в каждой древовидной структуре. На уровне стволов i имеется N i стволов, так что пусть j , j ∈ {1, …, N i } представляют все стволы на каждом уровне стволов. Мы также определяем ω ij как длину ствола j на уровне ствола i и определить χ ij , которое представляет количество листовых узлов, участвующих в стволе j на уровне ствола i .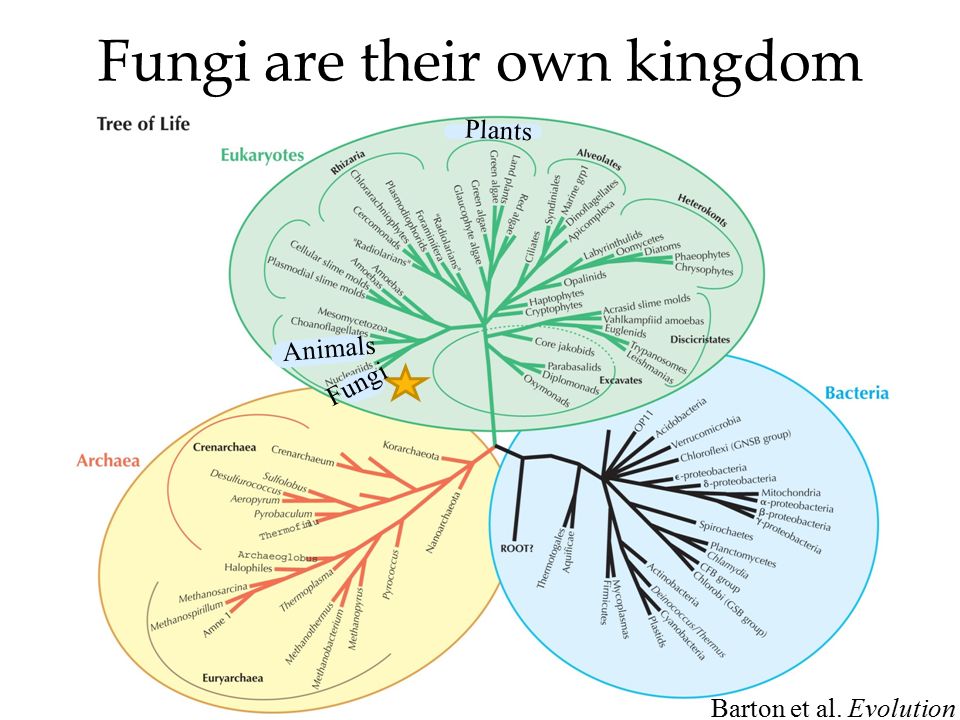 Тогда весовая оценка W T древовидной структуры T будет рассчитана по формуле: потому что древовидные структуры одного и того же пациента имеют один и тот же корневой ствол.
Тогда весовая оценка W T древовидной структуры T будет рассчитана по формуле: потому что древовидные структуры одного и того же пациента имеют один и тот же корневой ствол.
Аннотация мутаций-драйверов на филогенетическом дереве
Хорошо известно, что в геноме рака больше мутаций-пассажиров, чем мутаций-драйверов. Понимание времени возникновения и распределения драйверных мутаций в разных образцах важно для понимания эволюции опухолевой прогрессии. Поэтому наш метод также аннотирует предполагаемые 299 генов-драйверов на ветвях дерева для последующего анализа (Bailey et al., 2018). Следует отметить, что в древовидной структуре может быть более одной ветви дерева с аннотационной информацией одного и того же гена-драйвера. Это может быть вызвано одной и той же мутацией или разными мутациями одного и того же гена-драйвера, на что может ответить файл вспомогательной информации, соответствующий файлу древовидной структуры.
Поскольку этот метод предполагает, что для каждого образца существует один основной клон из-за нескольких биопсий, мы заметили, что в редких случаях этот метод будет выводить несколько решений вместо одного оптимального решения, когда некоторые образцы состоят из более чем одного основного клона ( Дополнительное примечание 3).
Результаты
Результаты при высокозлокачественном серозном раке яичников
Для оценки нашего метода мы сравнили эффективность PTI с двумя современными методами, LICHeE и Treeomics, при высокозлокачественном серозном раке яичников (HGSC). набор данных, которые были получены из Европейского архива геномов и феномов (инвентарный номер EGAS00001000547) (Bashashati et al., 2013). PTI использовала все мутации с AF >= 0,01, в то время как LICHeE и Treeomics запускали с параметром, определенным в их опубликованной статье. Затем мы сравнили результаты этих трех методов с результатами, приведенными в оригинальной литературе, основанными на мутациях и изменениях числа копий. Чтобы оценить сходство двух древовидных структур, мы определили систему оценки сходства древовидных структур. Показатель сходства представляет собой долю идентичных путей в топологии дерева и колеблется от нуля до единицы (дополнительное примечание 4). PTI показал несколько лучшую производительность по сравнению с LICHeE и Treeomics.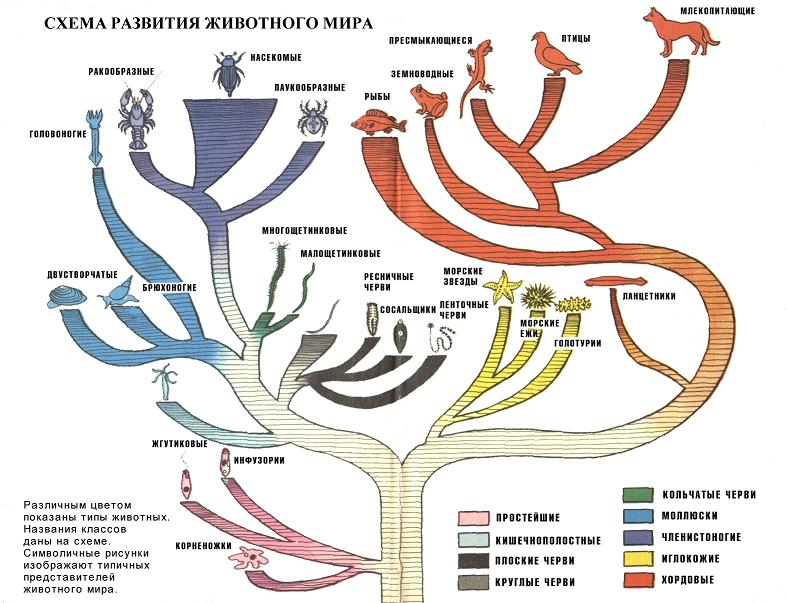 Только PTI правильно предсказал идентичную структуру в случае 4, где образцы j и f-i сгруппированы в одну ветвь (таблица 1 и рисунок 2A). 4 из 6 результатов, предсказанных PTI, показали идентичную структуру с показателем сходства, равным 1. Ни один из трех методов не показал высокосогласованных структур в случаях 1 и 5 в качестве исходных результатов (таблица 1 и рисунок 2B). В случае 1 результаты PTI и LICHeE были очень согласованными. Для случая 5, когда PTI использует все соматические мутации с AF >= 0,01, между древовидными структурами, полученными PTI и Treeomics, имеются лишь незначительные различия. Однако, если PTI использует все однонуклеотидные соматические мутации, полученные из исходной статьи, включая 8 образцов опухолей из случая 5, в отличие от результатов в исходной литературе, которые предполагают раннее расхождение образца c, PTI сначала отделяет образец h для достижения наибольшего общие мутации (n = 4) в остальных образцах (дополнительное примечание 5). Тщательный анализ набора данных о соматических мутациях показал, что если выборка с расходится первой, то в остальных выборках имеется только одна общая соматическая мутация.
Только PTI правильно предсказал идентичную структуру в случае 4, где образцы j и f-i сгруппированы в одну ветвь (таблица 1 и рисунок 2A). 4 из 6 результатов, предсказанных PTI, показали идентичную структуру с показателем сходства, равным 1. Ни один из трех методов не показал высокосогласованных структур в случаях 1 и 5 в качестве исходных результатов (таблица 1 и рисунок 2B). В случае 1 результаты PTI и LICHeE были очень согласованными. Для случая 5, когда PTI использует все соматические мутации с AF >= 0,01, между древовидными структурами, полученными PTI и Treeomics, имеются лишь незначительные различия. Однако, если PTI использует все однонуклеотидные соматические мутации, полученные из исходной статьи, включая 8 образцов опухолей из случая 5, в отличие от результатов в исходной литературе, которые предполагают раннее расхождение образца c, PTI сначала отделяет образец h для достижения наибольшего общие мутации (n = 4) в остальных образцах (дополнительное примечание 5). Тщательный анализ набора данных о соматических мутациях показал, что если выборка с расходится первой, то в остальных выборках имеется только одна общая соматическая мутация.
Таблица 1 Сравнение PTI, LICHeE и Treeomics на основе набора данных HGSC.
Рисунок 2 Древовидная структура, выведенная PTI для случаев 1, 4 и 5 HGSC. (A) Древовидная структура варианта 4, выведенная PTI, LICHeE и Treeomics соответственно. В результате ЛИЧЕ: желтая линия, внутренняя ветвь; голубая линия, ствол; черная линия, ссылка на вклад; числа внутри круга, SSNV; светло-фиолетовый квадрат, область опухоли. В результате Treeomics: числа, выделенные синим цветом, соответствуют полученным вариантам в ветвях. Проценты (серые) обозначают значения начальной загрузки (1000 выборок). СК, субклон. (B) Для случая 1 PTI сначала разделяет образец c для достижения наилучшего разделения ветвей, что соответствует результату LICHeE. Для случая 5 древовидная структура, полученная PTI и Treeomics, имеет большое сходство с использованием шести образцов и восьми образцов (дополнительное примечание 5).
Результаты по набору данных о светлоклеточном раке почки
Мы провели отдельное сравнение между PTI, LICHeE и BAMSE по набору данных по светлоклеточному раку почки (ccRCC) у восьми человек, которые были получены из Европейского архива генома и фенома (инвентарный номер EGAS00001000667). (Герлингер и др., 2014). Поскольку LICHeE использовала только частоту вариантных аллелей соматических однонуклеотидных вариантов для реконструкции филогенетического процесса, PTI взяла тот же набор SNV с AF >= 0,01 в качестве набора входных данных. Поскольку в этом наборе данных отсутствовала информация об общем количестве прочтений и вариантах прочтений мутаций (такая информация нужна Treeomics), мы сравнили результаты PTI, LICHeE, BAMSE только с результатами из оригинальной литературы, в которых использовалась кластеризация на основе VAF, шаблон присутствия вариантов и максимальная экономия. алгоритм (Gerlinger et al., 2014). Следует отметить, что деревья, полученные с помощью BAMSE, были получены от Toosi et al. (2019). Сравнение показывает, что PTI и LICHeE работали одинаково с точки зрения точности и скорости в наборе данных ccRCC, в то время как все древовидные структуры, выведенные BAMSE, имели различия в одной или нескольких ветвях (таблица 2, рисунок S7).
(Герлингер и др., 2014). Поскольку LICHeE использовала только частоту вариантных аллелей соматических однонуклеотидных вариантов для реконструкции филогенетического процесса, PTI взяла тот же набор SNV с AF >= 0,01 в качестве набора входных данных. Поскольку в этом наборе данных отсутствовала информация об общем количестве прочтений и вариантах прочтений мутаций (такая информация нужна Treeomics), мы сравнили результаты PTI, LICHeE, BAMSE только с результатами из оригинальной литературы, в которых использовалась кластеризация на основе VAF, шаблон присутствия вариантов и максимальная экономия. алгоритм (Gerlinger et al., 2014). Следует отметить, что деревья, полученные с помощью BAMSE, были получены от Toosi et al. (2019). Сравнение показывает, что PTI и LICHeE работали одинаково с точки зрения точности и скорости в наборе данных ccRCC, в то время как все древовидные структуры, выведенные BAMSE, имели различия в одной или нескольких ветвях (таблица 2, рисунок S7).
Таблица 2 Сравнение PTI и других методов на наборе данных ccRCC.
Результаты по набору данных о раке молочной железы
PTI также сравнивали с LICHeE и Treeomics по набору данных о раке молочной железы, а затем сравнивали эти результаты с результатами из оригинальной литературы, основанными как на соматических мутациях, так и на изменениях числа копий. Набор данных о раке молочной железы был получен из Европейского архива генома и фенома (инвентарный номер EGAS00001000760) (Brown et al., 2017). Время выполнения PTI, как и двух других методов, было коротким, всего в течение нескольких секунд (таблица S5). И PTI, и Treeomics показали более высокую точность по сравнению с LICHeE. В результатах, предсказанных PTI, 6 из 8 пациентов показали идентичные структуры, в то время как два других пациента P1 и P2 показали очень похожие древовидные структуры, как результаты Brown et al. (2017), что может быть вызвано более чем одной субклональной популяцией в одной биопсии (рис. 3). Например, у пациента P1 образец M1 (образец метастатической опухоли) включает A-клон и B-клон, образец M4 (образец метастатической опухоли) содержит только A-клон, а образцы M3 и P содержат B-клон. Поэтому образец M1 группируется вместе с образцом M4 или с образцами P-M3, что определяется долей соматических мутаций, вовлеченных в A-клон и B-клон в образце P (дополнительное примечание 6). Treeomics также показала хорошую производительность, и 6 из 8 результатов были идентичными. Однако по результатам LICHeE у 5 из 8 пациентов были обнаружены одноветвевые или многоветвевые различия в древовидных структурах. Мы также проверили эти два метода на разных частотах отсечки AF. Точность древовидных структур трех методов была немного улучшена, но PTI по-прежнему показал лучшую производительность (таблица S6). Кроме того, мы также продемонстрировали, что PTI надежно работает в наборе данных с низким охватом, применив его к набору данных HISEQ для 8 пациентов с раком молочной железы (таблица S7).
Поэтому образец M1 группируется вместе с образцом M4 или с образцами P-M3, что определяется долей соматических мутаций, вовлеченных в A-клон и B-клон в образце P (дополнительное примечание 6). Treeomics также показала хорошую производительность, и 6 из 8 результатов были идентичными. Однако по результатам LICHeE у 5 из 8 пациентов были обнаружены одноветвевые или многоветвевые различия в древовидных структурах. Мы также проверили эти два метода на разных частотах отсечки AF. Точность древовидных структур трех методов была немного улучшена, но PTI по-прежнему показал лучшую производительность (таблица S6). Кроме того, мы также продемонстрировали, что PTI надежно работает в наборе данных с низким охватом, применив его к набору данных HISEQ для 8 пациентов с раком молочной железы (таблица S7).
Рисунок 3 Сравнение деревьев для восьми больных раком молочной железы. Восемь филогенетических древовидных структур без аннотационной информации о гене-драйвере, полученном с помощью PTI с использованием всех соматических мутаций с охватом >=1500X и >=3% VAF (слева), сравниваются с деревьями (справа), опубликованными в Brown et al.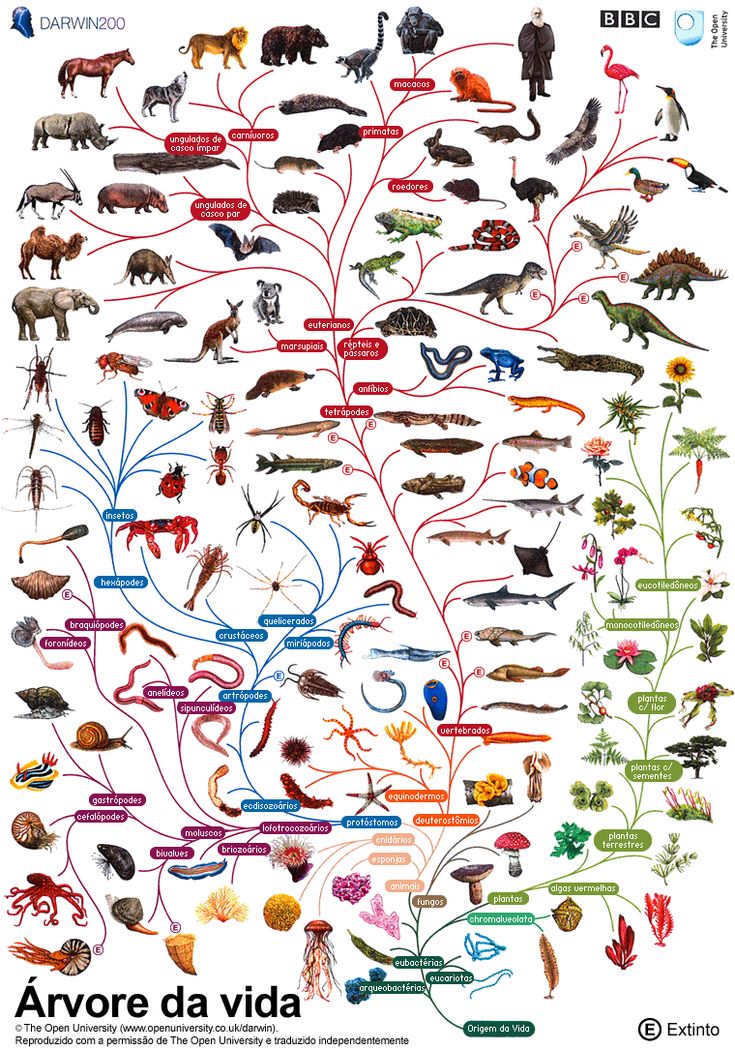 (2017) для каждого пациента в наборе данных о раке молочной железы. Что касается результатов PTI, количество общих мутаций пропорционально длине ветви и помечено над каждой ветвью. Кроме того, масштабные линейки в верхнем правом углу результатов, приведенных в оригинальной опубликованной статье, представляют 10 SNV и указывают исходную длину деревьев.
(2017) для каждого пациента в наборе данных о раке молочной железы. Что касается результатов PTI, количество общих мутаций пропорционально длине ветви и помечено над каждой ветвью. Кроме того, масштабные линейки в верхнем правом углу результатов, приведенных в оригинальной опубликованной статье, представляют 10 SNV и указывают исходную длину деревьев.
Результаты по 13 наборам данных о типах рака
Мы также провели PTI на реальном наборе данных, включая соматические однонуклеотидные мутации у 40 пациентов с 13 типами рака, для которых данные о частоте аллелей недоступны. Этот набор данных был получен из базы данных BioStudies (регистрационный номер S-EPMC4776530) (Zhao et al., 2016). Мы применили PTI для вывода древовидной структуры, а затем сравнили наши результаты с результатами, основанными на множественном выравнивании последовательностей, алгоритме максимального правдоподобия, алгоритме максимальной экономии и байесовских критериях вывода, реализованных в исходном исследовании. А затем, основываясь на показателе сходства, мы разделили результаты сравнения на четыре группы: показатель сходства = 1, показатель сходства ∈ [0,5,1), показатель сходства ∈ [0,2,0,5) и показатель сходства ∈ [0,0,2], представляющий различная степень сходства древовидной структуры. Сравнение показывает, что 92,5% наших результатов имеют такую же или аналогичную древовидную структуру (оценка сходства выше 0,2), что и результаты Zhao et al. (2016) (рис. 4, рис. S8), что еще раз свидетельствует о том, что наш метод может быть применим в широком диапазоне приложений.
А затем, основываясь на показателе сходства, мы разделили результаты сравнения на четыре группы: показатель сходства = 1, показатель сходства ∈ [0,5,1), показатель сходства ∈ [0,2,0,5) и показатель сходства ∈ [0,0,2], представляющий различная степень сходства древовидной структуры. Сравнение показывает, что 92,5% наших результатов имеют такую же или аналогичную древовидную структуру (оценка сходства выше 0,2), что и результаты Zhao et al. (2016) (рис. 4, рис. S8), что еще раз свидетельствует о том, что наш метод может быть применим в широком диапазоне приложений.
Рисунок 4 Краткий обзор эффективности нашего метода на наборе данных 13 типов рака без частоты аллелей. Соответствие между PTI и исходными результатами, основанное на оценках сходства, было представлено в виде круговой диаграммы.
Обсуждение
Поскольку PTI предполагает наличие одного основного клона в каждой биопсии, когда имеется более одного основного субклона, PTI относит образец к субклону с более высоким числом мутаций, а не к их относительной клеточной численности двух субклонов (рис. 3 и Таблица S4). Это может привести к некоторым расхождениям в древовидных структурах по сравнению с другими методами. Но этот случай редко наблюдается в исследованиях мультирегионального секвенирования, мы наблюдаем только один случай во всех проверенных нами случаях.
3 и Таблица S4). Это может привести к некоторым расхождениям в древовидных структурах по сравнению с другими методами. Но этот случай редко наблюдается в исследованиях мультирегионального секвенирования, мы наблюдаем только один случай во всех проверенных нами случаях.
В этом исследовании мы представляем PTI, новый и простой в использовании метод для определения филогенетического древа опухолевой прогрессии с использованием только соматических мутаций без необходимости глубокого секвенирования для получения высоконадежного измерения частоты аллелей. Наше сравнение с другими существующими методами, такими как LICHeE, Treeomics, BAMSE и другими традиционными методами, показывает, что PTI достигает аналогичной или немного лучшей производительности за короткое время, обычно менее минуты. Эта функция важна для изучения клинических образцов, в которых трудно получить точную информацию о частоте аллелей, таких как образцы, фиксированные формалином и залитые парафином (FFPE). Более того, входной файл для PTI представляет собой аналогичную матрицу с нулевым и одним признаком, так что этот метод в целом применим для вывода о филогении для любых других наборов данных, которые могут быть преобразованы в этот формат (например, эпигенетика). Фактически, этот метод также хорошо подходит для наборов данных отдельных клеток, чтобы оценить сходство между отдельными клетками и построить их филогении.
Фактически, этот метод также хорошо подходит для наборов данных отдельных клеток, чтобы оценить сходство между отдельными клетками и построить их филогении.
Заявление о доступности данных
В этом исследовании были проанализированы общедоступные наборы данных. Эти данные можно найти здесь: Европейский архив генома-фенома (EGAS00001000547, EGAS00001000667, EGAS00001000760) и биоисследования (S-EPMC4776530).
Вклад авторов
PW и LZ разработали алгоритм и интерпретировали результаты. PW реализовал алгоритм и провел анализ. LH помогал в получении и предварительной обработке наборов данных. YZ помог настроить программную среду для сравнения производительности.
Финансирование
Эта работа финансируется Национальной ключевой программой исследований и разработок Китая (2018YFC1004602), Национальным фондом естественных наук Китая (NSF 31871332) и стартовым фондом L.Z. из Шанхайского технологического университета.
Конфликт интересов
Авторы заявляют, что исследование проводилось при отсутствии каких-либо коммерческих или финансовых отношений, которые могли бы быть истолкованы как потенциальный конфликт интересов.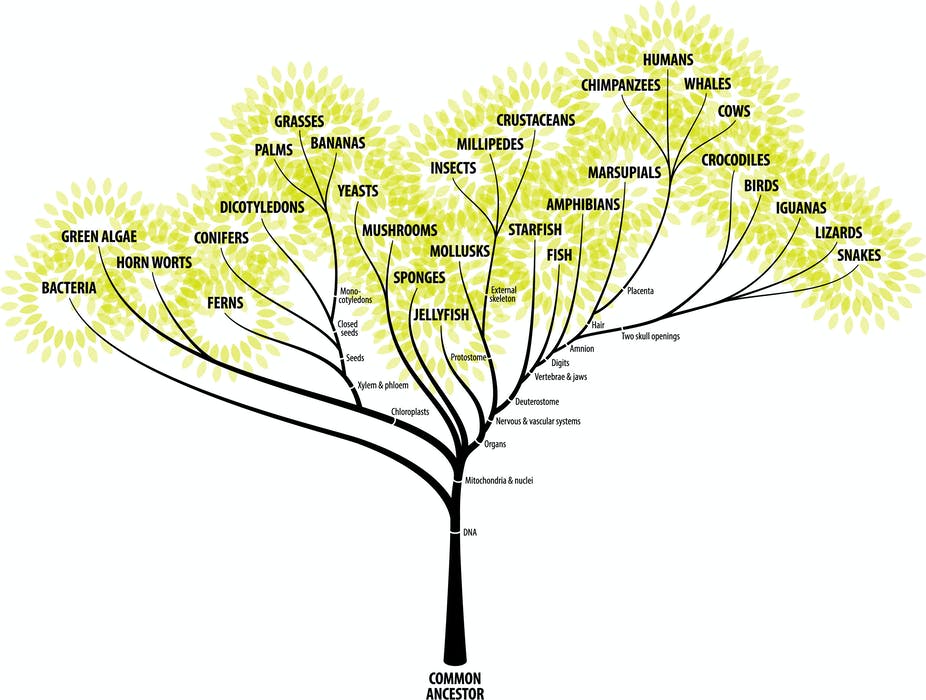
Благодарности
Мы хотели бы поблагодарить Xiuqi Pan за тестирование воспроизводимости данных. Мы хотели бы поблагодарить платформу высокопроизводительных вычислений Шанхайского технологического университета за поддержку.
Дополнительный материал
Дополнительный материал к этой статье можно найти в Интернете по адресу: https://www.frontiersin.org/articles/10.3389/fgene.2019.01371/full#supplementary-material
Ссылки
Astolfi, A. , Urbini, M., Indio, V., Nannini, M., Genovese, C.G., Santini, D., et al. (2015). Секвенирование полного экзома (WES) фиксированной формалином и залитой парафином (FFPE) опухолевой ткани при стромальных опухолях желудочно-кишечного тракта (GIST). BMC Genomics 16, 892. doi: 10.1186/s12864-015-1982-6
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Бейли, М. Х., Токхейм, К., Порта-Пардо, Э., Сенгупта, С., Бертран, Д., Вирасинг, А., и др. (2018). Всесторонняя характеристика генов-возбудителей рака и мутаций. Ячейка 173, 371–385 e318. doi: 10.1016/j.cell.2018.02.060
Ячейка 173, 371–385 e318. doi: 10.1016/j.cell.2018.02.060
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Башашати А., Ха Г., Тон А., Дин Дж., Прентис Л. М., Рот А. и др. (2013). Различные эволюционные траектории первичного высокозлокачественного серозного рака яичников, выявленные с помощью пространственного мутационного профилирования. Дж. Патол. 231, 21–34. doi: 10.1002/path.4230
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Браун Д., Смитс Д., Секели Б., Ларсимонт Д., Сас А. М., Аднет П. Ю. и др. (2017). Филогенетический анализ метастатического прогрессирования рака молочной железы с использованием соматических мутаций и аберраций числа копий. Нац. коммун. 8, 14944. doi: 10.1038/ncomms14944
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Чой, Ю. Дж., Ри, Дж. К., Хур, С. Ю., Ким, М. С., Ли, С. Х., Чанг, Ю. Дж., и др. (2017). Внутрииндивидуальная геномная гетерогенность серозной карциномы яичника высокой степени злокачественности и клиническая полезность асцитных раковых клеток для профилирования мутаций. Дж. Патол. 241, 57–66. doi: 10.1002/path.4819
Дж. Патол. 241, 57–66. doi: 10.1002/path.4819
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Цибульскис К., Лоуренс М. С., Картер С. Л., Сиваченко А., Яффе Д., Сугнез К. и др. (2013). Чувствительное обнаружение соматических точечных мутаций в неочищенных и гетерогенных образцах рака. Нац. Биотехнолог. 31, 213–219. doi: 10.1038/nbt.2514
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Ферроника П., Хоф Дж., Кац-Угурлу Г., Сеймонс Р. Х., Терпстра М. М., Де Ланге К. и др. (2019). Всестороннее профилирование первичного и метастатического скПКР выявляет высокую гомологию метастазов субрегиону первичной опухоли. Раков 11, 812. doi: 10.3390/cancers11060812
CrossRef Полный текст | Google Scholar
Герлингер М., Роуэн А. Дж., Хорсвелл С., Ларкин Дж., Эндесфельдер Д., Гронрус Э. и др. (2012). Внутриопухолевая гетерогенность и разветвленная эволюция выявлены с помощью мультирегионального секвенирования. Новый англ. Дж. Мед. 366, 883–892. дои: 10.1056/NEJMoa1113205
Новый англ. Дж. Мед. 366, 883–892. дои: 10.1056/NEJMoa1113205
Полнотекстовая перекрестная ссылка | Google Scholar
Герлингер М., Хорсвелл С., Ларкин Дж., Роуэн А. Дж., Салм М. П., Варела И. и др. (2014). Геномная архитектура и эволюция светлоклеточных почечно-клеточных карцином, определенная многорегиональным секвенированием. Нац. Жене. 46, 225–233. doi: 10.1038/ng.2891
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Gundem, G., Van Loo, P., Kremeyer, B., Alexandrov, L.B., Tubio, J.M.C., Papaemmanuil, E., et al. (2015). Эволюционная история летального метастатического рака предстательной железы. Природа 520, 353–357. doi: 10.1038/nature14347
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Хонг, М. К. Х., Макинтайр, Г., Ведж, Д. К., Ван Лоо, П., Патель, К., Лунке, С., и др. (2015). Отслеживание источников и факторов субклональной метастатической экспансии при раке предстательной железы. Нац. коммун. 6, 6605. doi: 10.1038/ncomms7605
Нац. коммун. 6, 6605. doi: 10.1038/ncomms7605
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Цзяо В., Вембу С., Дешвар А. Г., Штейн Л., Моррис К. (2014). Вывод клональной эволюции опухолей из однонуклеотидных соматических мутаций. BMC Биоинф. 15, 35. doi: 10.1186/1471-2105-15-35
CrossRef Полный текст | Google Scholar
Kim, T.M., Jung, S.H., An, C.H., Lee, S.H., Baek, I.P., Kim, M.S., et al. (2015). Субклональные геномные архитектуры первичного и метастатического колоректального рака на основе внутриопухолевой генетической гетерогенности. клин. Рак рез. 21, 4461–4472. doi: 10.1158/1078-0432.CCR-14-2413
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Кобольдт Д. К., Чжан К., Ларсон Д. Э., Шен Д., Маклеллан М. Д., Лин Л. и др. (2012). VarScan 2: обнаружение соматических мутаций и изменений числа копий при раке путем секвенирования экзома. Рез. генома. 22, 568–576. doi: 10.1101/gr.129684.111
doi: 10.1101/gr.129684.111
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Lu, Y.W., Zhang, H.F., Liang, R., Xie, Z.R., Luo, HY, Zeng, Y.J., et al. (2016). Генетическая гетерогенность колоректального рака, определенная с помощью многорегионального секвенирования. PloS One 11, e0152673. doi: 10.1371/journal.pone.0152673
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Маликич С., Макферсон А.В., Донмез Н., Сахиналп К.С. (2015). Вывод о клональности в нескольких образцах опухолей с использованием филогенеза. Биоинформатика 31, 1349–1356. doi: 10.1093/bioinformatics/btv003
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Марусик А., Альмендро В., Поляк К. (2012). Внутриопухолевая гетерогенность: зазеркалье для рака? Нац. Преподобный Рак 12, 323–334. doi: 10.1038/nrc3261
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Naxerova, K. , Reiter, JG, Brachtel, E., Lennerz, J.K., Van De Wetering, M., Rowan, A., et al. (2017). Происхождение лимфатических и отдаленных метастазов при колоректальном раке человека. Наука 357, 55–60. doi: 10.1126/science.aai8515
, Reiter, JG, Brachtel, E., Lennerz, J.K., Van De Wetering, M., Rowan, A., et al. (2017). Происхождение лимфатических и отдаленных метастазов при колоректальном раке человека. Наука 357, 55–60. doi: 10.1126/science.aai8515
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Nieboer, M.M., Dorssers, L.C.J., Straver, R., Looijenga, LHJ, De Ridder, J. (2018). TargetClone: подход с несколькими образцами для реконструкции субклональной эволюции опухолей. PloS One 13, e0208002. doi: 10.1371/journal.pone.0208002
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Попик В., Салари Р., Хаджирасулиха И., Кашеф-Хагиги Д., Уэст Р. Б., Бацоглу С. (2015). Быстрый и масштабируемый вывод мультивыборочных линий рака. Геном Биол. 16, 91. doi: 10.1186/s13059-015-0647-8
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Reiter, JG, Makohon-Moore, A.P., Gerold, JM, Bozic, I., Chatterjee, K. , Iacobuzio-Donahue, C.A., et al. (2017). Реконструкция метастатических моделей рака человека. Нац. коммун. 8, 14114. doi: 10.1038/ncomms14114
, Iacobuzio-Donahue, C.A., et al. (2017). Реконструкция метастатических моделей рака человека. Нац. коммун. 8, 14114. doi: 10.1038/ncomms14114
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Сато Ф., Саджи С., Той М. (2016). Геномная эволюция опухоли рака молочной железы. Рак молочной железы 23, 4–11. doi: 10.1007/s12282-015-0617-8
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Шварц Р.Ф., Нг, С.К.Ю., Кук С.Л., Ньюман С., Темпл Дж., Пискорц А.М. и др. (2015). Пространственная и временная неоднородность при высокозлокачественном серозном раке яичников: филогенетический анализ. ПлоС Мед. 12, е1001789. doi: 10.1371/journal.pmed.1001789
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Туси, Х., Моейни, А., Хаджирасулиха, И. (2019). BAMSE: выбор байесовской модели для вывода о филогении опухоли среди нескольких образцов. BMC Биоинф. 20, 282. doi: 10. 1186/s12859-019-2824-3
1186/s12859-019-2824-3
CrossRef Full Text | Google Scholar
Йейтс, Л. Р., Кэмпбелл, П. Дж. (2012). Эволюция генома рака. Нац. Преподобный Жене. 13, 795–806. doi: 10.1038/nrg3317
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Йейтс Л. Р., Герстунг М., Кнаппског С., Десмедт К., Гандем Г., Ван Лоо П. и др. (2015). Субклональная диверсификация первичного рака молочной железы, выявленная мультирегиональным секвенированием. Нац. Мед. 21, 751–759. doi: 10.1038/nm.3886
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Йейтс Л. Р., Кнаппског С., Ведж Д., Фармери Дж. Х. Р., Гонсалес С., Мартинкорена И. и др. (2017). Геномная эволюция метастазов и рецидивов рака молочной железы. Раковая клетка 32, 169–184 e167. doi: 10.1016/j.ccell.2017.07.005
PubMed Abstract | Полный текст перекрестной ссылки | Google Scholar
Zhai, W., Lim, T.K., Zhang, T., Phang, S.T., Tiang, Z.